Monocarboxylate transporter 8 is an important thyroid hormone transporter
Thyroid hormone (TH) is essential for the normal growth and development of many tissues, particularly the brain. TH is the common name for the prohormone thyroxine (3,3’,5,5’-tetraiodo-L-thyronine, T4), which is the predominant product secreted by the thyroid gland, and for the active hormone T3 (3,3’,5-triiodo-L-thyronine), which is largely produced in peripheral tissues by deiodination of T4. Since T3 receptors and the catalytic sites of the deiodinating enzymes are located intracellularly, TH has to cross the cell membrane. For this process, transmembrane transporter proteins are required.1–3 Over the last decade, multiple TH transporter proteins have been identified and characterized,4 of which the monocarboxylate transporter 8 (MCT8) is the only transporter currently linked to a clinical syndrome. MCT8 facilitates cellular influx and efflux of T4 and T3 and, to a lesser extent, rT3 (3,3’,5’-triiodo-L-thyronine) and 3,3’-T2 (3,3’-diiodo-L-thyronine), which designates MCT8 as the most specific TH transporter identified to date.5,6 The gene encoding MCT8 (SLC16A2) is located on chromosome Xq13.2. MCT8 is expressed in many tissues including the thyroid, liver, kidney, vascular endothelial cells, and the brain.4 Its expression in the vascular endothelial cells at the blood–brain barrier (BBB), as well as in oligodendrocytes and neuronal cells is of critical importance to maintain adequate TH homeostasis in the brain during different developmental stages. This is illustrated by patients with a defect in MCT8 function, resulting in the Allan– Herndon–Dudley syndrome (AHDS).7,8
The clinical characteristics of the Allan–Herndon–Dudley syndrome
The clinical phenotype of AHDS is dominated by two key features: a severe neurocognitive phenotype and signs of peripheral thyrotoxicosis (see Table 1).
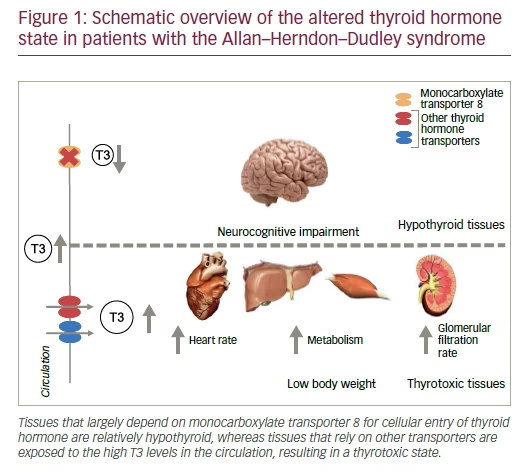
Given its critical role in the maintenance of adequate intracerebral TH levels, the current paradigm holds that mutations in MCT8 result in a hypothyroid state of the brain, compromising normal brain development (see Figure 1). This results in a severe neurocognitive phenotype, which is dominated by a moderate to severe delay in cognitive development combined with central hypotonia, resulting in an evident head-lag, inability to sit and maintain postural balance without support, and frequent episodes of dystonic and dyskinetic movements and hypertonia of the extremities.8–12 The latter has long been confused with spasticity, which only becomes apparent with increasing age in the extremities (it is more pronounced in lower extremities) as is evidenced by the occurrence of hyperreflexia, hypertonia, tightening of the heel cords and clonus.9 Moreover, a positive Babinski sign is often present.9 In addition to these key aspects, the absence of speech development and the presence of (choreo)athetosis, swallowing difficulties, drooling, acquired microcephaly and seizures have been frequently reported.8–13 Small-scale magnetic resonance imaging studies in relatively young AHDS patients (<10 years) have shown that gross morphology

is often normal.9,12,14,15 A prominent delay in myelination is the most consistent finding.9,12,14,15 However, the progression of myelination to (near) normal in older subjects, which can be especially observed in the corpus callosum and corticospinal tracts, may help to differentiate AHDS from other white matter disorders such as leukodystrophies, cerebral palsy and Pelizaeus-Merzbacher disease.9,16,17 Diffusion tensor imaging (DTI) may show a poor definition of predominantly the anteroposteriorly directed white matter association tracks, which is consistent with the presence of dystonia.9
In addition to the neurocognitive phenotype, patients suffer from severe peripheral thyrotoxicosis as a result of high serum T3 levels (see Figure 1).7,8,18 Interestingly, FT4 and TT4 levels are low or low-normal, rT3 levels are frequently very low and thyroid-stimulating hormone (TSH) levels are often high-normal. Together these abnormalities in TFTs constitute an endocrine fingerprint for the presence of AHDS in a male patient with a severe intellectual disability. These observations suggest that mutations in MCT8 directly affect TH metabolism and the integrity of the hypothalamic– pituitary–thyroid axis, which is supported by multiple animal studies.19–21 In particular, increased T4 accumulation in the thyroid gland, trapping of T4 in the kidney and a pronounced increase in D1 activity in peripheral tissues seem to contribute to the abnormal serum TH levels in animal models.22,23 Not surprisingly, most patients suffer from thyrotoxic symptoms such as (very) low body weight, increased perspiration, tachycardia and muscle wasting. Since adequate dietary intake is already compromised by the severe neurological phenotype, the increase in basal metabolism as a result of high T3 levels can result in life-threatening cachexia.
As a result of its X-linked inheritance, AHDS predominantly affects males. In rare cases, AHDS may also present in females as a result of chromosomal translocations and unfavourable non-random X-inactivation in female carriers.24 Although the majority of female carriers appear asymptomatic, the occurrence of minor symptoms in female carriers has never been systematically studied and thus remains speculative. 25
The challenge of diagnosing the Allan–Herndon– Dudley syndrome
The presence of the characteristic abnormal TFT pattern is suggestive for AHDS, although a similar endocrine fingerprint with neurocognitive features can occur in patients with TRá1-mutations.26 Nevertheless, the diagnostic process has proven to be very challenging in many AHDS patients. This is illustrated by the advanced age at which most currently known patients have been diagnosed. Several factors contribute to this problem.
First of all, AHDS is a rare genetic disorder, which intrinsically results in lower awareness among healthcare professionals.27 Moreover, due to its low prevalence, clinical expertise is scattered throughout the world, with most doctors only having one family under their care. As a result, awareness, knowledge and expertise often remain isolated. Moreover, the predominant neurological features (for example, persistent hypotonia) early in the disease course are not always linked to abnormal TH signaling.
Secondly, key clinical characteristics that differentiate AHDS from other neurodevelopmental disorders are poorly defined. This is primarily caused by the large variation in the severity of the clinical phenotype of AHDS patients, which may be related to the differential impact of mutations on MCT8 function. Whereas most patients fail to reach any major motor milestones, some patients are able to stand and walk without support and have gained speech development. Although many studies have reported on the phenotypic characteristics of the AHDS, most of them describe only a small number of patients, often within the same family. Moreover, these studies often focus on different aspects, using non-uniform evaluation methods. This greatly hampers the comparison of patients between studies and thus the identification of core signs and symptoms that may trigger the determination of serum TFTs. Moreover, it is important to measure serum (F)T3 levels in addition to the routinely performed (F)T4 and TSH measurements to avoid misdiagnosis. The combination of low (F)T4 and normal TSH is often misinterpreted for secondary hypothyroidism, further delaying the diagnostic process.
Thirdly, AHDS is not detected with the current neonatal screening programs. In most countries, neonatal screening programs for congenital hypothyroidism (CH) are based on measurement of TSH levels alone or in combination with T4 levels, while in a small number of countries the measurement of T4 levels is the first step of the screening procedure. Either way, patients with AHDS are not likely to be detected based on the combination of their T4 and TSH levels, since T4 levels are only mildly decreased, while TSH levels are often within the normal range. Theoretically, AHDS patients could be detected by implementing a T3 measurement in the screening procedure. However, it remains to be explored to what extent T3 levels are already abnormal in the neonatal period.
Currently available therapeutic options
The therapeutic approach of AHDS should at least contain two important modalities. The first modality consists of supportive care and reduction of symptomatology including physical, occupational, and speech therapies, medical treatment of dystonia and seizures (if present) and most importantly the maintenance of a positive energy balance by dietary supplementation. In particular, the medical treatment of dystonia and seizures is empirical. Further studies into the exact underlying pathogenic mechanisms may allow more targeted treatment approaches. Moreover, frequent orthopaedic follow-up is required to monitor the development and progression of scoliosis.
The second modality entails the restoration of adequate TH signalling and should aim to:
• restore the hypothyroid state in tissues that largely rely on MCT8 (MCT8- dependent tissues) for their cellular TH uptake; and
• protect tissues that mostly rely on other TH transporters (MCT8- independent tissues) from the high serum T3 levels that result in a thyrotoxic state (see Figure 1).
Prompted by the low serum FT4 and TT4 and high normal to slightly elevated TSH levels, which were interpreted as mild hypothyroidism, many AHDS patients have been empirically treated with LT4. However, in general LT4 administration does not result in any clinical improvement and even leads to a further increase in serum (F)T3 levels aggravating the thyrotoxic symptoms.28–32 Therefore, LT4 monotherapy should be discouraged in AHDS patients.
In contrast, LT4 administration in combination with propylthiouracil (PTU) has been shown to effectively normalize serum TFTs leading to a significant increase in body weight.27,33 However, as expected, no changes in the neurocognitive phenotype have been observed, since LT4 requires functional MCT8 to enter the brain. Moreover, long-term administration of PTU harbours a risk of severe side-effects, such as agranulocytosis and liver failure. Although LT4 in combination with PTU is currently the best treatment strategy available in routine patient care to restore the abnormal TFTs, it is still far from ideal. The restoration of adequate TH signalling in the brain cannot be achieved using this combination therapy, prompting the search for alternative therapeutic agents.
Therapeutic options under investigation
In order to restore TH levels in the MCT8-dependent, hypothyroid tissues, an effective therapeutic agent should
• circumvent MCT8 for its cellular uptake;
• act like T3 inside the cell; and
• be metabolized like T3 to allow natural fine-tuning of TH signalling.
Several T3 analogues have been subject of extensive pre-clinical studies, of which the synthetic T3 analogue diiodothyropropionic acid (DITPA) and the naturally occurring T3 metabolite Triac have shown the most promising results.34–38 DITPA has been shown to effectively restore serum T3 levels in Mct8 knock-out mice.35,36 Administration of DITPA to four AHDS patients initiated at 8.5–25 months resulted in normalization of serum T3 levels and improvement of some markers that reflect the TH state in peripheral tissues.37 However, no consistent changes in body weight or neurocognitive phenotype were observed. Triac holds strong potential as low concentrations enabled dendritogenesis in cultured Purkinje cells.38 Of note, Triac administration shortly after birth was shown to promote normal brain development in Pax-8 knockout (KO) and MCT8/Oatp1c1 double KO mice.34 The effects of Triac in AHDS patients are currently under investigation in a clinical trial (NTC02060474).
Although the effects of T3 analogues on the peripheral thyrotoxicosis seem promising, it remains to be explored to what extent the neurocognitive phenotype in human patients can be modulated by these compounds once treatment is initiated postnatally. Obviously, the highest therapeutic potential can be expected if treatment is initiated as early as possible after birth or, theoretically, in a prenatal context. Interestingly, several (pre-clinical) studies have shown that both DITPA and Triac cross the placenta, which may allow treatment of the unborn foetus in utero in case of a pre-natal diagnosis.39,40
Future directions in the Allan–Herndon–Dudley syndrome treatment
Pre-clinical studies are currently directed to explore the potential use of chemical chaperones, in order to restore the expression of functional MCT8.41 Given the monogenetic cause of AHDS, gene therapy could be an alternative solution to restore functional MCT8 expression. Further studies are warranted to explore the effect and feasibility of these therapeutic approaches.
Conclusion
Early detection of AHDS is essential to enable early treatment initiation. Clinicians should be aware that AHDS cannot be ruled out based on a ‘normal’ neonatal screening result for CH. Therefore, we advise to measure not only serum (F)T4 and TSH, but also (F)T3 and, if available, rT3 levels, in all male subjects with moderate-severe delay in cognitive and motor development. Given the severity of the disease, it is paramount to carefully evaluate each potential therapy. As for other rare diseases, it is crucial to join forces in the development of treatment strategies for AHDS by sharing and centralizing clinical expertise.