Pituitary adenomas are benign tumours that arise from the adenohypophysis. They are the second most frequent intracranial tumour type after meningiomas, and account for 16.2% of all primary cranial neoplasms.1 Though likely an underestimate, the incidence of pituitary adenomas is approximately four per 100,000 persons per year,2,3 and incidence increases with age.4 Prolactinomas and non-functioning pituitary adenomas are the most common pituitary adenoma types, followed by somatotroph, corticotroph and thyrotroph adenomas. Almost all gonadotroph adenomas are clinically non-functioning, and less than 1% are hormonally active.4,5 Functioning pituitary adenomas carry significant morbidity and increased mortality due to resultant clinical syndromes, concurrent hypopituitarism as well as tumour mass effect.6–8 Prompt and effective treatment is crucial to disease control and a reduction in associated health risks.9–11 In this review, treatment options for different types of functioning pituitary adenomas are presented, with a focus on current and emerging medical therapies.
Pituitary adenoma therapy
Therapy for pituitary adenomas includes transsphenoidal surgery, medical treatment and/or radiation therapy.
Surgery
Transsphenoidal surgery is the first-line therapy for most cases of functioning pituitary adenomas (except prolactinomas), as surgery can achieve rapid and sustained biochemical remission, along with decompression of the optic chiasm. However, surgery carries the risk of new pituitary deficiencies (3.6–19.4%), transient or permanent diabetes insipidus (4.3–17.7% and 0.3–7.3% respectively), hyponatremia (4.3–21%) and other surgical complications such as cerebrospinal fluid (CSF) leak (2.6–7%), haemorrhage (1.1–2.9%), infection (1.1–3.8%), carotid artery injury (0.1–1.1%) and vision loss (0.6–1.8%).12–14 In a meta-analysis, hypopituitarism was more common after transsphenoidal surgery for Cushing’s disease (25%) than acromegaly, prolactinoma or non-functioning pituitary adenomas (approximately 7–12%), and is thought to be related to prolonged glucocorticoid replacement and a more aggressive surgical technique used for corticotroph adenomas.15 Rates of surgical success and complications vary, with more favourable outcomes achieved with experienced surgeons and in high-volume centres.13,16 Microscopic and endoscopic techniques appear equally effective, with a similar complication rate.12,14
Medical
Medical therapy is generally used as adjunct therapy after a failed transsphenoidal surgery in Cushing’s disease and acromegaly, when surgery cannot be performed, or for recurrent disease. Often medical therapy becomes a long-term treatment option that requires monitoring for biochemical control and side effects. Pituitary-directed medications (somatostatin receptor ligands [SRLs] and dopamine agonists) exert antisecretory and antiproliferative effects on pituitary tumours; prolactinomas typically respond to dopamine agonists with considerable tumour regression, while in acromegaly and Cushing’s disease, tumour response to SRLs varies significantly. End-organ targeted therapy, such as inhibitors of adrenal steroidogenesis or receptor blockers, e.g. glucocorticoid or growth hormone receptor antagonists, can provide effective biochemical and/or clinical disease control (Table 1).
Radiation
Due to slow onset of response (up to several years) and development of new pituitary deficiencies, radiation therapy is considered a third-line therapy option following unsuccessful transsphenoidal surgery and failed medical treatment, or in cases of tumour recurrence.10,11 Conventional and stereotactic fractionated radiation is delivered in multiple small doses over 5–6 weeks, while stereotactic radiosurgery is performed in a single-session, high-dose treatment. Conventional and stereotactic radiosurgery therapy have similar effectiveness, although stereotactic radiosurgery may result in quicker normalisation of hypersecreted hormones.17–19 Stereotactic radiosurgery is more convenient for a patient; however, it carries a higher risk of damage to the optic apparatus in tumours located very close to the optic chiasm.18–20 Secondary brain tumours develop in 1.1–2.4% of patients after conventional therapy with relative risk ranging from no increased risk to 10.5 compared to the general population, and several studies have shown no increased risk compared to nonirradiated patients.21,22 Conventional radiotherapy may pose a slightly higher risk of stroke, especially in acromegaly,23 and it has been associated with neurocognitive impairment, specifically in verbal memory and executive function, regardless of tumour type.24
Prolactinomas
Prolactinomas are the most common functioning pituitary tumours (66%), with a female:male ratio of 10:1.2,25 The majority are microadenomas (80%) and men more commonly present with macroadenomas.2,26 Giant prolactinomas represent 1–5% of all prolactinomas.27
Medical
Dopamine agonists, the mainstay of medical therapy, are very effective at normalising prolactin levels and reducing pituitary adenoma size; resulting in a rapid, often within days, visual improvement and gradual restoration of hypogonadism and fertility.28,29 Cabergoline and bromocriptine are dopamine agonists available in the United States. Another dopamine agonist, quinagolide, is used in some European countries. Cabergoline is recommended over bromocriptine due to higher potency and effectiveness when compared with bromocriptine.28 Cabergoline normalises prolactin in 83% versus bromocriptine in 59% of women with hyperprolactinaemic amenorrhea.30 A meta-analysis of six observational studies and three randomised trials showed superiority of cabergoline at reducing prolactin levels and associated symptoms of hypogonadism in women.31 The effect on tumour volume reduction has not been assessed in randomised controlled trials. However, data extracted from two separate studies suggest greater efficacy of cabergoline on tumour volume reduction, 96% versus 64% with bromocriptine.32–34 This difference in efficacy is thought to be related to cabergoline having a stronger affinity for dopamine D2 receptors and a longer duration of action.28
Resistance, defined as inability to achieve normoprolactinaemia or 50% reduction in tumour volume with standard doses of dopamine agonist, occurs in 25% of patients treated with bromocriptine and 10% treated with cabergoline.28,35 It is estimated that up to 80% of those resistant to bromocriptine can achieve normal prolactin levels with cabergoline.28 The mechanism of resistance is also poorly understood, but may be due to decreased number of D2 receptors, presence of different receptor isoforms or downstream signalling changes in resistant prolactinomas.28,36,37 Cabergoline dose may be increased, typically (1–2 mg/week) to a maximal tolerable dose, though this is rarely reported to overcome resistance.28
Common side effects of dopamine agonists are nausea, dizziness and headache, with nausea being more pronounced in patients treated with bromocriptine. Additionally, there is growing evidence of an association of dopamine agonist use with impulse control disorders such as pathologic gambling and hypersexuality.38 Although the prevalence of these disorders in patients with prolactinomas is still unknown, one study reported that the risk of developing an impulse control disorder was 9.9 times higher in males treated with dopamine agonists compared to those with non-functioning adenomas.38,39 A very rare but serious complication is CSF leak in giant prolactinomas eroding the sellar floor, which typically requires urgent neurosurgical intervention.40 Although standard cabergoline doses are not associated with increased risk of valvular heart disease, doses >2 mg/week and high cumulative doses may still carry some risk, and monitoring with echocardiograms has been recommended by some groups.41–45 Few studies have reported association of bromocriptine with non-clinically significant valve fibrosis,46,47 and larger studies have reported no association.43
Investigation of emerging medical therapies is underway (Table 2). Lapatinib is a tyrosine-kinase inhibitor of epidermal growth factor receptor (EGFR) and receptor tyrosine-protein kinase (ErbB2 or human epidermal growth factor receptor 2 [HER2]), which shows promise as a treatment for resistant prolactinomas. Lapatinib has been demonstrated to decrease prolactin levels by 60% and 40% in transgenic mice with pituitary expression of EGFR and HER2, respectively; prolactin remained unaltered in control mice.48 In two human subjects, addition of lapatinib to cabergoline after prolonged treatment with high dose cabergoline allowed for 22% tumour volume reduction and almost normalisation of prolactin at 6 months in one case, and suppression of tumour growth with 42% prolactin reduction in the other case.49,50 Side effects were mild alopecia, rash, diarrhoea and anorexia. Lapatinib is currently being evaluated in the phase II trial, Targeted Therapy with Lapatinib in Patients with Recurrent Pituitary Tumors Resistant to Standard Therapy (ClinicalTrials.gov Identifier; NCT00939523).
Surgery
Transsphenoidal surgery is reserved for patients with resistant prolactinomas, those with intolerance or contraindications to medical therapy, as well as emergent situations such as apoplexy and CSF leak. Several studies have examined the outcomes of transsphenoidal surgery and reported variable remission rates; range 30–93%,51 with lower remission rates reported for invasive prolactinomas. Following transsphenoidal surgery, new pituitary deficiencies developed in 17.1% of patients in one study (7.0% anterior pituitary hypofunction), while improvement of existing deficiencies occurred in 14.6%.52 Although prolactinomas in men tend to be more aggressive, it has not been clearly demonstrated that men have poorer surgical outcomes.26,53 However, a recent study of prolactinomas in males who required surgery reported a high rate of residual tumour (92.6%) and frequent need for additional surgery and radiation.26 Preoperative dopamine-agonist therapy does not appear to significantly affect surgical cure.26,51,53 Following debulking surgery, medical therapy normalises prolactin levels in almost half of resistant adenomas, and with lower dopamine agonists doses.51 Recurrence of hyperprolactinaemia after initial remission following transsphenoidal surgery is common, reportedly varying between 5–58%; higher rates are reported in studies with longer follow up.51,52,54
Table 1: Current medical options for prolactinoma, acromegaly and Cushing’s disease
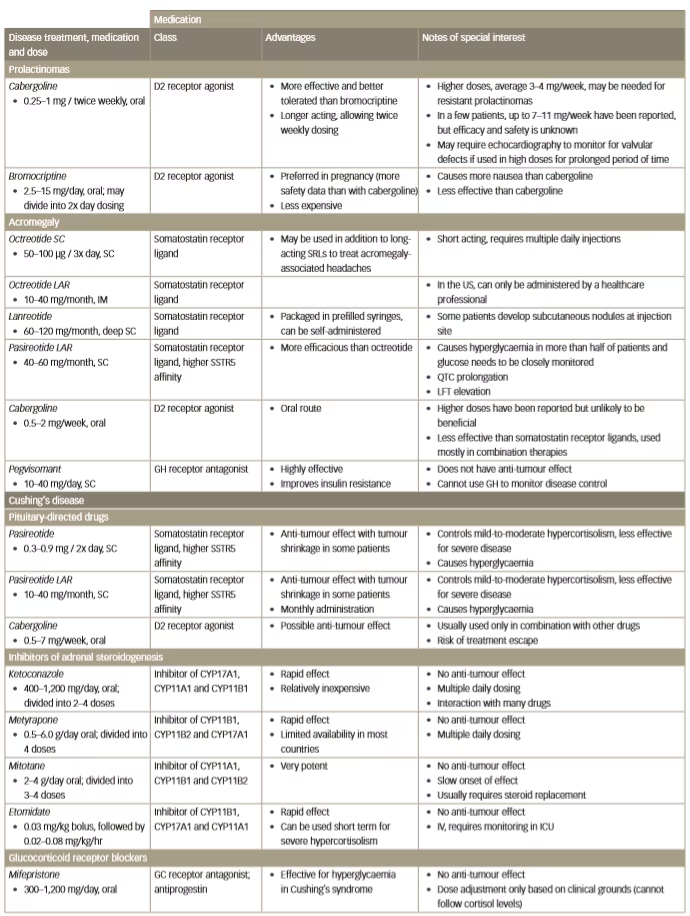
D = diarrhoea; D2 = dopamine receptor D2; GC = glucocorticoid; GH = growth hormone; ICU = intensive care unit;IM = intramuscular; IV = intravenous; LFT = liver function test; SRL = somatostatin receptor ligand; SSTR5 = somatostatin receptor 5; SC = subcutaneous; V = vomiting. QTC = electrocardiogram calculated duration of time from the start of the Q wave to the end of the T wave adjusted for a patient’s heart rate.
Table 2: Emerging investigational drug medical therapies for pituitary adenomas
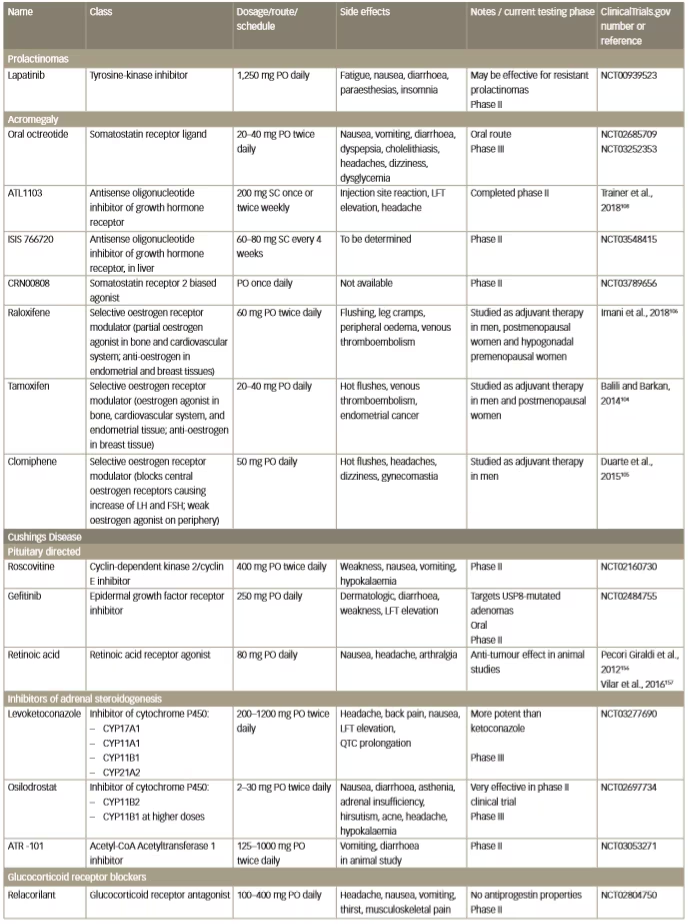
FSH = follicle stimulating hormone; LFT = liver function test; LH = luteinizing hormone; PO = per so (by mouth); SC = subcutaneously.
QTC = electrocardiogram calculated duration of time from the start of the Q wave to the end of the T wave adjusted for a patient’s heart rate.
Radiation
Radiation therapy is used for resistant and aggressive or malignant prolactinomas.55 Prolactin levels normalise in 26–52% of patients post radiation therapy, tumour growth is controlled in 89–92%55–57 and hypopituitarism occurs in one-third of patients.55 Some authors have reported that dopamine agonist use at the time of radiation may reduce remission rates, while others did not find this association.55,58,59 Therefore, there is no universal recommendation to withhold dopamine agonists prior to radiation.
Acromegaly
Growth hormone-producing adenomas represent approximately 9.0–13.2% of pituitary adenomas; 73.0% are macroadenomas,2,5,60 and giant adenomas constitute about 4.5%.27 Acromegaly affects males and females equally and is most commonly diagnosed during the fifth decade of life.61
Surgery
Transsphenoidal surgery is recommended as the first-line treatment due to rapid control of growth hormone levels. When performed by an experienced surgeon, remission is greater than 85% for microadenomas62,63 and 40–66% for macroadenomas.63,64 If biochemical remission is not achieved, patients require adjunctive medical therapy and/or radiation therapy. Even when the likelihood of cure is low, such as in the case of large or invasive adenomas, debulking surgery is still recommended as it provides better postoperative control by SRLs.65–8 Five-year recurrence after surgery is reported as 0.7–5.4% in various studies, and 10–15-year recurrence as 0.1–10%.64,69–71 Incidence of pituitary deficiencies after transsphenoidal surgery include 6.50% for adrenal insufficiency, 4.39% for central hypothyroidism, 6.70% for hypogonadism, 14.95% for growth hormone deficiency and 10.05% for transient and 2.42% for permanent diabetes insipidus, as assessed by a meta-analysis.72
Overall, surgery is more effective than primary medical therapy for treatment-naïve patients. A meta-analysis of prospective and retrospective studies demonstrated that surgically treated patients had higher remission rates than their medically treated counterparts (67% versus 45%, p=0.02).73
Finally, surgery can provide additional information about tumour granulation type and presence of somatostatin receptor (SSTR) on pathology. Densely granulated adenomas and SSTR2A-positive adenomas respond better to SRLs than sparsely granulated adenomas and SSTR2A-negative adenomas.74,75 This knowledge allows for an SRL response prediction, which serves to guide physicians in selecting individualised patient treatment plans (e.g. SRL monotherapy versus pegvisomant or early combination therapy).
Medical
Medical therapy may be used as primary therapy in poor candidates for surgery, in those who decline surgery or if surgery is unlikely to provide biochemical cure due to extent of the disease.10 Occasionally, medical therapy is used preoperatively in high-risk patients with a goal of reducing growth hormone levels and decreasing anaesthesia complications such as laryngeal oedema, high-output heart failure and uncontrolled hypertension.76,77 SRLs, growth hormone receptor antagonists (pegvisomant) and dopamine agonists are the three classes of medications currently used to treat acromegaly. They are used as a single agent or in combination.7,78
SRLs act on SSTR subtypes 2 and 5 located on somatotroph cells. First-generation SRLs (octreotide [OCT] long-acting release [LAR], lanreotide slow release and lanreotide autogel) seem to be equally effective in controlling growth hormone and insulin-like growth factor 1 (IGF-1) level in approximately 55% of patients (reported clinical studies range 17–70%).10,79,80 The wide range in reported efficacy can be explained by different clinical study methodology, selection bias, previous surgery or medical treatment, dose, duration of follow-up and other factors.79 Biochemical control can be maximised by escalating the dose of OCT-LAR (60 mg every 4 weeks)81 and lanreotide (180 mg every 4 weeks or 120 mg every 3 weeks) in treatment-responsive patients.82–84 First-generation SRLs are effective at shrinking the tumour in 30–66% of patients by 10–77%.85–87 Approximately one-third of patients experience 50% tumour volume reduction with primary medical therapy.86 Common side effects include gastrointestinal distress, gallstones and dysglycaemia.
Pasireotide is a multi-SRL with a high affinity for SSTR5 compared to other SRLs and has been demonstrated to have higher efficacy than OCT-LAR in a randomised controlled trial (31.3% versus 19.2%, respectively, p=0.007), while tumour volume reduction was similar, approximately 40%.88 Additionally, pasireotide can normalise biochemical control in 20% of patients resistant to first-generation SRLs;89 however, hyperglycaemia occurs in 57% of patients. Pasireotide-induced hyperglycaemia has been attributed to a reduction in insulin secretion, glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide levels.90 Close monitoring in patients with impaired glucose tolerance and diabetes is required and treatment with metformin, dipeptidyl peptidase-4 (DPP-4) inhibitor or GLP-1 receptor agonist has been recommended for the management of hyperglycaemia.91
Pegvisomant is a highly selective growth hormone receptor antagonist that blocks growth hormone receptor to reduce IGF-1 production by the liver. Pegvisomant is used in patients who remained uncontrolled after transsphenoidal surgery or radiation therapy and sometimes as primary therapy if transsphenoidal surgery is not feasible or desired.10,92 In clinical trials, pegvisomant effectiveness was remarkable, with IGF-1 normalisation rates of up to 97% on maximal doses of 40 mg daily.93 However, long-term surveillance studies indicated that a real-world control rate was approximately 60%, likely reflecting lower standard doses used in everyday practice compared with clinical trials.10,82 Liver function tests are monitored and treatment should be discontinued if transaminases increase to greater than five-times the upper limit of normal (ULN). Despite a theoretical concern that lack of IGF-1 feedback may stimulate pituitary tumour growth, progression of tumour was noted only in 3% of patients.94 Although this may represent natural tumour progression independent of pegvisomant, serial magnetic resonance imaging (MRI) has been recommended.10
Dopamine agonists normalise IGF-1 and growth hormone in one-third of patients,95,96 and induce tumour volume reduction in up to 62%.95 Due to modest efficacy in acromegaly, cabergoline plays a role mostly as adjunctive therapy in cases with mildly elevated IGF-1.
Combination medical therapy
Pegvisomant can be used in combination with first-generation SRLs in patients who are uncontrolled on SRL alone. This may result in
IGF-1 normalisation in up to 95% of cases.97 A combination of pegvisomant and pasireotide may potentially allow for lower doses.
This has been examined in one study to date,98,99 where it was demonstrated that switching from first-generation SRLs to pasireotide allowed for a reduction in pegvisomant dose by half. Pegvisomant did not offset or prevent pasireotide-induced hyperglycaemia. Diarrhoea was a common side effect, but no elevation of transaminases was observed.
Dopamine agonists in combination with SRLs achieve normalised IGF-1 levels in approximately half of patients who are uncontrolled on SRLs alone.95 Cabergoline plus a low-dose pegvisomant combination is also a modestly effective option when liver enzyme elevation, diabetes or SRL-induced hyperglycaemia is a concern.100
Oestrogen is capable of lowering IGF-1 through inhibition of liver growth hormone receptor expression or through upregulation of suppressors of cytokine signalling-2, leading to decreased growth hormone signalling.101,102 Selective oestrogen receptor modulators bind to oestrogen receptors and exhibit oestrogen agonistic or antagonistic effects in different tissues. Raloxifene, tamoxifen and clomiphene have been studied as an add-on therapy in men and post-menopausal women with uncontrolled acromegaly.103–106 In one study, tamoxifen normalised IGF-1 in eight (47%) patients and decreased IGF-1 in 14 (82%) patients.104 A recent head-to-head open label study of raloxifene versus cabergoline add-on therapy to long-acting SRL showed similar lowering effect on IGF-1; IGF-1 normalised in 45.5% of patients with raloxifene and in 40.9% of patients with cabergoline.106 Adverse effects included flushing. A study of clomiphene in men has shown a decrease in IGF-1 by 41%, including normalisation of IGF-1 in 44% without reported side effects.105 Selective oestrogen receptor modulators are not part of standard acromegaly management and longer-term studies are needed to assess their safety and efficacy.106
Emerging medical therapy
Several new acromegaly treatment agents as well as a new SRL formulations are being tested in human clinical trials (Table 2). In a phase III clinical trial, oral OCT maintained biochemical control in 62% of patients who were switched from a long-acting SRL (from 89% controlled on SRL at baseline).107 There are two other ongoing phase III trials: Comparison of Oral Octreotide Capsules to Injectable Somatostatin Analogs in Acromegaly (MPOWERED; ClinicalTrials.gov Identifier: NCT02685709) and Efficacy and Safety of Octreotide Capsules (MYCAPSSA) in Acromegaly (OPTIMAL; ClinicalTrials.gov Identifier: NCT03252353).
An antisense oligonucleotide inhibitor of growth hormone receptors is a novel and promising therapy. A phase II study of ATL1103 demonstrated a 27.8% reduction of IGF-1 at week 14, with good tolerability but mild-to-moderate injection-site reaction occurring in 85%.108 A newer generation antisense oligonucleotide ISIS 766720, targeting hepatic expression of growth hormone receptor, is currently being investigated in a phase II trial (Safety, Tolerability, and Efficacy of IONIS-GHR-LRx in up to 42 Adult Patients with Acromegaly Being Treated with Long-acting Somatostatin Receptor Ligands; ClinicalTrials.gov Identifier: NCT03548415).
Another emerging therapeutic agent is an orally bioavailable nonpeptide SSTR2 biased agonist, CRN00808. CRN00808 is biased for SSTR2 activation (a process that causes reduction of growth hormone secretion) over receptor internalisation (which is a process that limits therapeutic activity of SSTR agonist). Preliminary phase I clinical trial data (Single and Multiple-Ascending Dose Study of CRN00808 in Healthy Volunteers; ClinicalTrials.gov Identifier: NCT03276858) show that a 2.5 mg once-daily dose lowers growth hormone-releasing hormone-induced growth hormone release by 73% in healthy volunteers.109 A phase II study evaluating CRN00808 in patients with acromegaly treated with SRLs is ongoing (A Study to Evaluate the Safety and Efficacy of CRN00808 for the Treatment of Acromegaly [ACROBAT EDGE]; ClinicalTrials.gov Identifier: NCT03789656).
Radiation
Radiation therapy is reserved for patients with persistent disease after surgery who failed medical therapy. However, remission onset is slow, taking many years and requiring medical therapy in the interim. Hormonal remission rates are 50–60% at 10–15 years with both stereotactic radiosurgery and conventional radiation, with stereotactic radiosurgery slightly more effective.23,110,111 Hypopituitarism develops in up to 50% of patients at 5 years.23,110 Some suggested that SRLs may prevent a full effect of radiation, but this has not been a consistent observation and may be due to the fact that in non-randomised studies, patients with more severe disease were more likely to continue SRL prior to treatment.20,112,113 Although not clearly recommended by current guidelines, some centres withhold SRLs for four to eight weeks prior to radiation therapy.20
Cushing’s disease
Adrenocorticotropic hormone (ACTH)-producing adenomas represent 4–6% of all adenomas, occurring more frequently in females (3:1 ratio).2,5 Only about a third are macroadenomas, while the majority are microadenomas, and approximately 12% of those are not detectable on MRI.5,114 Aggressive corticotroph adenomas are usually biochemically silent, and have an approximately 30% risk of recurrence.115
Surgery
Transsphenoidal surgery is the recommended treatment of choice for ACTH-producing adenomas. Remission rates range from 65–98%, with higher rates when a pituitary adenoma is identified on MRI and removed completely by an experienced and specialised surgeon.116,117 If a pituitary adenoma is not found during surgical exploration,
hemi-hypophysectomy/pan-hypophysectomy may be performed, with remission rates reported as 60–75%.116 However, more extensive transsphenoidal surgery carries greater risk of hypopituitarism. Postoperative pituitary deficiencies develop in 25% of patients with Cushing’s disease compared to approximately 7–13% in other pituitary adenomas.15 Transient diabetes insipidus is the most common postoperative pituitary dysfunction occurring in 4–48% of patients, while permanent diabetes insipidus has been reported in 3–46% of patients.15,118 Rates of postoperative thyrotropin and gonadotropin deficiency are 11–20% and 8–17% respectively.118–120 In one study, prevalence of growth hormone deficiency was 65% in patients who achieved long-term surgical remission.121
Failure of remission occurs when a pituitary adenoma is incompletely excised or missed on surgical exploration, if dural invasion is present, or if a pituitary adenoma is extra-pituitary.116,122,123 Repeat surgery is an option for those with persistent hypercortisolism; however, remission rates are lower, 57–71%.124,125 Given the possibility of delayed remission, the decision to repeat surgery should be postponed until persistent disease is biochemically confirmed. Hypercortisolism can recur in up to 35% of cases.116,126
Medical
Medical therapy is necessary for persistent or recurrent hypercortisolism after transsphenoidal surgery or if surgery is contraindicated or declined. Additionally, medical therapy can be used post-radiation therapy until radiation effect occurs. Current medical therapy is directed at three targets: ACTH production by a corticotroph tumour, steroidogenesis in the adrenal gland, and glucocorticoid receptors.
Pituitary-directed drugs include pasireotide and cabergoline. Pasireotide binds to SSTR5 receptor, which is predominantly expressed on corticotroph adenoma cells, and inhibits ACTH production. In a 12-month clinical trial of pasireotide in Cushing’s disease, subcutaneous pasireotide decreased urinary free cortisol (UFC) by approximately 50% and normalised levels in more than 20% of patients, mainly in those with mild-to-moderate cortisol hypersecretion. Tumour volume reduction was 43% on a maximum dose of 900 µg daily.127 A long-acting monthly intramuscular formulation, pasireotide LAR, is available and has a similar efficacy as the subcutaneous pasireotide.128 The most frequent side effect of both formulations is hyperglycaemia, occurring in more than half of patients. Gastrointestinal side effects such as nausea, diarrhoea and cholelithiasis are similar in frequency to other SRLs.127 Cabergoline acts via D2 receptors on cortocotroph tumours. It is mostly used as an add-on therapy. Short-term response with UFC normalisation is observed in 25–35% of patients,129–131 while long-term efficacy is lower, mainly due to treatment escape.129,132 The dose range is 1–7 mg/week, with a commonly used dose of 2–3.5 mg/week.
Currently available inhibitors of steroidogenesis are ketoconazole, metyrapone, mitotane and etomidate. Ketoconazole is an antifungal agent used off-label in the United States (licensed in Europe) for Cushing’s syndrome. Ketoconazole inhibits cytochrome P450 enzymes on multiple levels of steroidogenesis and at higher doses effectively lowers glucocorticoid and androgen synthesis.133 Studies report efficacy in 30–90% of patients. The largest retrospective study to date showed that 49% of patients with Cushing’s disease achieved normal UFC and 25% had at least 50% reduction in UFC; however, 15% experienced escape from control after 2 years of treatment.134–7 Severe ketoconazole-induced hepatotoxicity (black box warning in the United States) is rare; however, mild liver enzyme elevation is relatively common and therefore requires close monitoring.138 Gynecomastia and hypogonadism due to inhibition of androgen synthesis limits use in males.
Metyrapone can effectively lower cortisol in 43–76% of patients with Cushing’s syndrome without apparent escape.139 Blockade of 11-beta hydroxylase causes accumulation of mineralocorticoid and androgenic precursors resulting in hypertension, hypokalaemia, oedema, hirsutism and acne. Mild gastrointestinal symptoms are common. Mitotane plays a major role in the treatment of adrenal carcinoma due to its adrenolytic effect on tumour cells, but it is occasionally used in Cushing’s disease. Very potent, but with a slow onset of action, mitotane induces eucortisolaemia in 72–82% of patients11 and often leads to adrenal insufficiency, requiring hydrocortisone replacement. Side effects include gastrointestinal upset, lethargy and abnormal liver function. Etomidate is a parenteral anaesthetic, which at lower, sub-hypnotic doses, rapidly inhibits cortisol production and thus has been used in patients with severe Cushing’s syndrome in the acute setting as well as preoperatively to decrease the risk of hypercortisolaemia related complications.140
Mifepristone is a glucocorticoid receptor antagonist that has been shown to significantly improve clinical manifestations of Cushing’s syndrome, including glucose metabolism, hypertension and weight gain. Overall, a clinical response was observed in up to 87% of patients.141 However, its mechanism of action causes ACTH and cortisol levels to rise during treatment, and therefore, they cannot be used to guide management; caution is required for potential tumour enlargement, especially in the case of macroadenomas.142 When encountered, adrenal insufficiency due to glucocorticoid receptor blockade should be treated with high doses of dexamethasone. Additionally, unopposed mineralocorticoid activity of cortisol can cause hypertension, oedema and hypokalaemia. The latter can be ameliorated by use of the mineralocorticoid blocker spironolactone. Also, an antiprogestin effect may result in endometrial hyperplasia.143
Combination medical therapy
Combination therapy is increasingly used for those patients who have failed monotherapy or if side effects of a single agent do not permit a dose increase. Regimens such as pasireotide plus cabergoline, ketoconazole plus cabergoline, and ketoconazole plus metyrapone have been successfully utilised.144,145
Emerging medical therapy
Multiple new medical therapies are currently in development and are undergoing clinical testing (Table 2). New inhibitors of steroidogenesis include levoketoconazole, osilodrostat and ATR-101. Levoketoconazole is a more potent enantiomer of ketoconazole and was proposed to achieve similar effectiveness with smaller doses and less pronounced side effects than ketoconazole. In a phase III, open-label trial, 81% of patients had initial UFC normalisation at the end of the dose titration period (up to 21 weeks), but only 30% of patients had normal UFC levels during the maintenance phase without a dose increase.146 Nausea and headache were most common side effects and liver function test elevation of >3 ULN in 11% of patients is relatively similar to the ketoconazole data from retrospective and observational data, although the studies are not comparable.139 Levoketoconazole is currently being assessed in a phase III, double-blind, withdrawal and rescue/restoration study (A Study to Assess the Safety and Efficacy of Levoketoconazole in the Treatment of Endogenous Cushing’s Syndrome; ClinicalTrials.gov Identifier: NCT03277690).
Osilodrostat is an oral nonsteroidal corticosteroid biosynthesis inhibitor that inhibits 11 beta-hydroxylase with higher affinity than metyrapone and has a longer half-life.147 In an open label study, osilodrostat induced normalisation of UFC in 84% of patients at week 10 and 79% at week 22.148 Despite the increase in the precursor 11-deoxycorticosterone, hypokalaemia was mild and hypertension has not been observed. Hirsutism and acne occurred in one-third of women due to elevated testosterone. Two phase III studies are ongoing: Safety and Efficacy of LCI699 for the Treatment of Patients with Cushing’s Disease (ClinicalTrials.gov Identifier: NCT02180217) and Efficacy and Safety Evaluation of Osilodrostat in Cushing’s Disease (LINC-4) (ClinicalTrials.gov Identifier: NCT02697734).
ATR-101, is a selective acyl-coenzyme A:cholesterol acyltransferase 1 (ACAT1)1 inhibitor that reduces cholesterol ester formation from cholesterol, thereby decreasing the substrate supply for steroidogenesis in the adrenal glands. ATR-101 was shown to reduce cortisol level by 50% in animal studies and is currently being studied in a phase II, double-blind study in humans (A Study of ATR-101 for the Treatment of Endogenous Cushing’s Syndrome; ClinicalTrials.gov Identifier: NCT03053271).
Relacorilant (CORT125134) is a selective antiglucocorticoid receptor antagonist that has an advantage of avoiding the progesterone receptor inhibitory effects of mifepristone. A phase II trial is ongoing (Study to Evaluate CORT125134 in Patients with Cushing’s Syndrome,
ClinicalTrials.gov Identifier: NCT02804750). Preliminary results of the low-dose arm showed improvement in glucose control, blood pressure, marker of bone formation and, somewhat surprisingly, a relatively small increase in ACTH and cortisol.149
Pituitary-targeted therapies in phase II trials include roscovitine and gefitinib. Roscovitine is a cyclin-dependent kinase inhibitor that suppresses 2/cyclin E on corticotroph tumour cells causing inhibition of the ACTH precursor hormone, proopiomelanocortin (POMC), with a subsequent decrease of ACTH production, although with a minimal antiproliferative effect.150 It is currently studied in a dose of 400 mg orally twice daily for 4 days every week, for a total of 4 weeks in Cushing’s disease (Treatment of Cushing’s Disease with R-roscovitine; ClinicalTrials.gov Identifier: NCT02160730).
Roscovitine has also been assessed in phase I trials in patients with various malignant solid tumours; some showed stabilisation of growth.151,152 Side effects were dose dependent and included fatigue, skin rash, hyponatremia and hypokalaemia occurring at doses of 800 mg and higher.151 Gefitinib is an EGFR inhibitor approved for non-small cell lung cancer. EGF binds to EGFR on corticotroph tumour cells and promotes POMC and ACTH synthesis.152 Approximately half of corticotroph tumours harbour a somatic USP8 mutation which causes over-expression of EGFR.153 Gefitinib has been shown to inhibit ACTH production in USP8-mutated corticotroph tumours.76,152 A study of gefitinib 250 mg once daily for 4 weeks is ongoing (Targeted Therapy with Gefitinib in Patients with USP8-mutated Cushing’s Disease; ClinicalTrials.gov Identifier: NCT02484755). Common adverse events are rash, diarrhoea and elevation of liver enzymes.153 Interstitial lung disease of grade 3–4 occurs in 0.7% of patients (package insert).154 Other mutations harboured by corticotroph tumours (e.g. USP48 and BRAF) are being actively investigated, but are beyond the scope of this review.155
Retinoic acid decreases ACTH by suppressing pro-opiomelanocortin gene transcription and exerts an anti-tumour effect in corticotroph adenomas.156 In a small prospective study, retinoic acid at 80 mg daily for 6–12 months decreased UFC to 22–73% of baseline levels; however, some patients were non-responders.156 Another small study (16 patients) showed a normalisation of UFC in 25% of participants at study end.157 Emerging pituitary-targeted drug treatments not yet in clinical trials include heat shock protein inhibitors, histone deacetylase inhibitors, monoclonal ACTH antibodies and others.158,159
Radiation
Radiation therapy is used when pituitary surgery is unsuccessful, in invasive adenomas or in poor surgical candidates. All patients should receive medical treatment while awaiting radiation effects, which may take several years. Control of hypercortisolism is achieved in up to 83% of patients undergoing conventional radiation and 70% undergoing stereotactic radiosurgery.18,160 Time to remission with stereotactic radiosurgery appears to be shorter, 14.5 months versus 18.0–42.0 months with conventional radiation.161 Recurrence after initial control has been observed in 18% of patients after stereotactic radiosurgery, which confirms the need for lifelong monitoring.161
Bilateral adrenalectomy
Bilateral adrenalectomy is reserved as a third-line treatment option for patients with uncontrolled hypercortisolism despite pituitary surgery, appropriate medical therapy and/or pituitary radiation. It can be life-saving in patients with severe and prolonged Cushing’s syndrome who require rapid and permanent control of hypercortisolism.162,163 Lastly, it may be selected earlier in patients desiring fertility options in whom pituitary irradiation or surgery is likely to result in irreversible hypogonadism. Rarely, hypercortisolism may persist or recur due to remnant adrenal tissue. Corticotroph tumour progression (Nelson’s syndrome) occurs in 0–47% of patients post bilateral adrenalectomy,164,165 and some data suggest that prior pituitary radiation may reduce the risk.166,167 It is important to periodically monitor ACTH and pituitary MRI. Mortality in the first year seems to be high post bilateral adrenalectomy.164
Thyroid stimulating hormone-secreting adenomas
Thyroid stimulating hormone (TSH)-secreting adenomas are rare, 0.5–3.0% of all pituitary adenomas, with equal male–female distribution, and are mostly macroadenomas at diagnosis.168 Surgical resection is the first-line treatment, with remission rates of 55–83%.169,170 Complete surgical removal is often impossible due to the fibrotic nature of these tumours and parasellar extension/invasion, necessitating adjunct medical therapy and/or radiation.168 Pre-treatment with antithyroid drugs, beta-blockers and SRLs is necessary in some cases to control severe hyperthyroidism to reduce the risk of perioperative thyroid storm.169,171 In a series of 68 operated patients (67% macroadenomas), six patients (9%) developed new hypopituitarism; hypogonadism in two, hypoadrenalism in two, both hypoadrenalism and hypogonadism in one, and panhypopituitarism in one patient.169 A smaller series of 13 patients reported postoperative hypopituitarism in five patients (38%); hypoadrenalism in three, hypothyroidism in one, growth hormone deficiency in two and hypogonadism in one patient.172 Radiation for persistent disease controls hyperthyroidism in 20–50% and produces tumour volume reduction in 26% of patients, and hypopituitarism occurs in approximately one-third of patients.169
Octreotide and lanreotide reduce TSH and normalise thyroid hormone levels in more than 80% patients.169,173 In a study of pre-operative OCT-LAR for a median of 33.5 days, T4 normalisation occurred in 84% of patients.172 Another study of mixed patient population (post pituitary surgery with or without additional radiation or medical therapy and those who were treatment-naïve) showed that lanreotide was effective at normalising thyroid hormone levels in 81% of patients.174 Resistance and escape from therapy are uncommon. As marked suppression of thyroid function can occur, patients should be monitored for hypothyroidism. SRLs induce tumour shrinkage in 39–61% of patients.171,174 Although TSH-secreting tumours express D2 receptors, dopamine agonists are less effective, with favourable results observed mostly in mixed prolactin/TSH-secreting adenomas.168,175
Gonadotroph adenomas
Functioning gonadotroph adenomas are very rare and treatment algorithms are limited to case reports and small case series. Adenomectomy is, however, the principal treatment approach; it restores gonadal function in men and women, and leads to the resolution of ovarian and testicular enlargement caused by hyperstimulation by gonadotroph adenoma.176 Radiation has been used as adjunct therapy after surgery, as well as for recurrent adenomas.177–180 Dopamine agonists, SRLs, gonadotropin-releasing hormone agonists and antagonists have been tried both as primary therapy and after transsphenoidal surgery, and in the majority of cases showed no benefit for clinical symptoms or tumour shrinkage.176
Aggressive pituitary adenomas
Aggressive pituitary adenomas are those that exhibit clinically significant growth despite appropriate medical, surgical and radiation therapy.181 They are often characterised by an elevated Ki68 index (≥3–10%), high p53 immunoreactivity and increased number of mitoses; however, these criteria have not been validated as strong prognostic markers of aggressiveness. Debulking surgery or near-total resection is recommended, especially for tumours with mass-effect on optic chiasm. Radiation therapy following surgery should be considered in patients without contraindication to radiation therapy, as it can provide a durable control of tumour growth. Medical therapy usually includes the use of temozolomide, an alkylating agent, alone or in combination with standard medical therapy for functioning pituitary adenomas (dopamine agonist, SRLs).182,183 Temozolomide is typically administered in cycles of 5 days every 28 days, with a maximum number of cycles of 9–12. Response rate (tumour shrinkage) differs by tumour type and is estimated from a small case series to be 44% in prolactinomas, 56% in corticotroph tumours, 38% in somatotroph tumours and only 22% in non-functioning tumours.182 Absent or low expression of O6-methylguanine-DNA methyltransferase, a DNA repair enzyme that interferes with temozolomide action, has been correlated with better response to temozolomide by some, but not all, authors.184,185 Temozolomide is usually well tolerated; however, myelosuppression can occur.185
Conclusion
Management of functioning pituitary adenomas often requires multiple treatment modalities to achieve rapid and durable remission and thus improve morbidity, mortality and quality of life. Treatment should be tailored individually based on the tumour type, availability of each therapeutic option and patient preference. Emerging medical therapies are being developed and may present future suitable options for patients with uncontrolled disease or intolerance to other medications.⬛