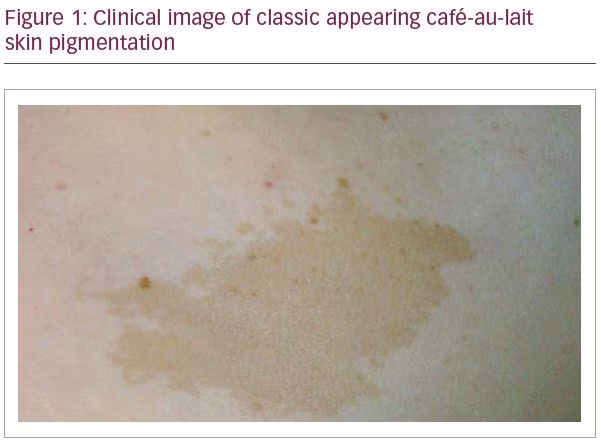
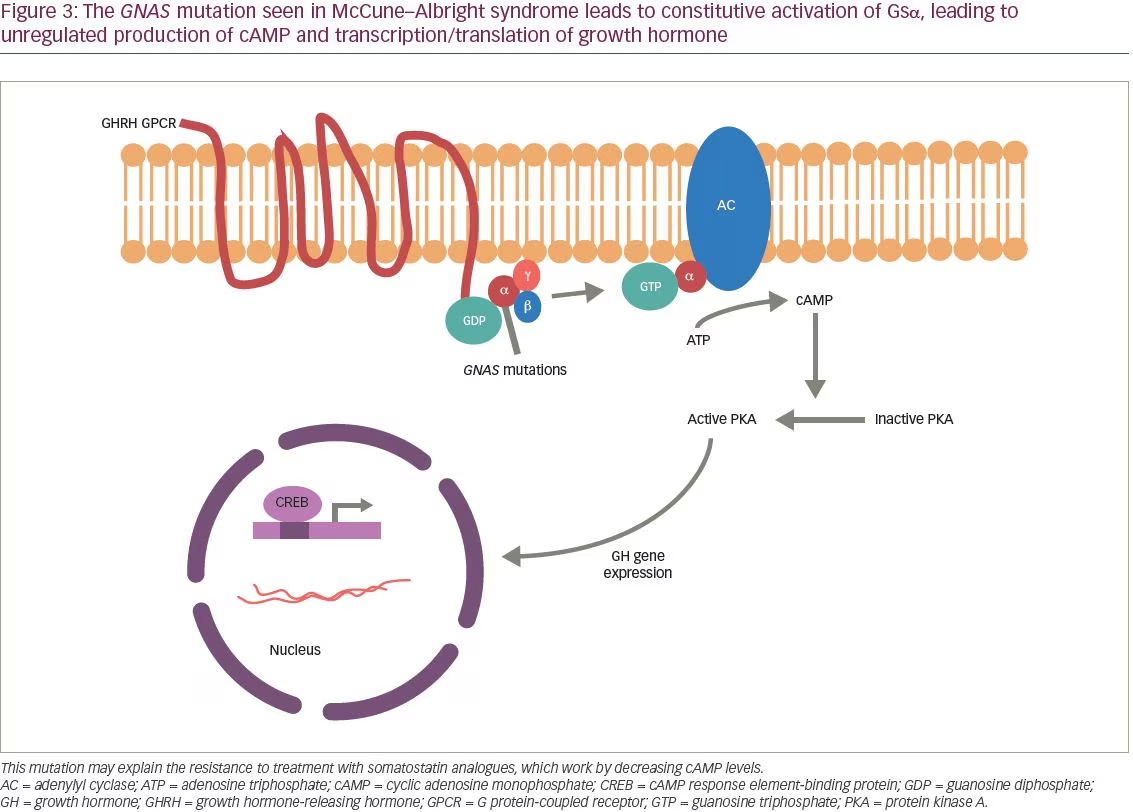
McCune–Albright syndrome (MAS) is a genetic disorder first described by McCune1 and Albright2 in the 1930s as a triad of polyostotic fibrous dysplasia, café-au-lait spots, and precocious puberty (Figure 1). Since then, other hyperfunctioning endocrinopathies have been described in MAS, including hyperthyroidism, Cushing’s syndrome, hypophosphatemic osteomalacia, and acromegaly.3 Patients with MAS display mosaicism as the disease results from a postzygotic mutation in the GNAS gene. GNAS is located on chromosome 20q13, which encodes for the stimulatory G protein alpha subunit (Gsa). The mutation causes constitutive activation of Gsα, leading to constitutive activation of adenylate cyclase and generation of cyclic adenosine monophosphate (cAMP).4 The distribution of affected cells plays a significant role in patient presentation, with about 20% of patients with MAS presenting with acromegaly.5
Treatment of growth hormone (GH) secreting pituitary adenomas in patients with MAS remains a challenge due to severe fibrous dysplasia of the skull base preventing access to the sella and the dangers of malignant transformation as a result of radiotherapy. As a result, medical management of MAS-associated acromegaly has been recommended as safe and effective.6 Somatostatin analogs and dopamine receptor agonists have traditionally been used, but often lead to only partial symptom and disease control.3,7 More recently, Akintoye et al. showed that pegvisomant, a direct GH receptor antagonist, is an effective alternative in patients with MAS who are unresponsive to somatostatin analogs.3 Below, we describe a case of a 15-year-old girl with MAS who was a poor surgical candidate and in whom somatostatin analogs were used to achieve normalized insulin-like growth factor 1 (IGF-1) levels.
Case presentation
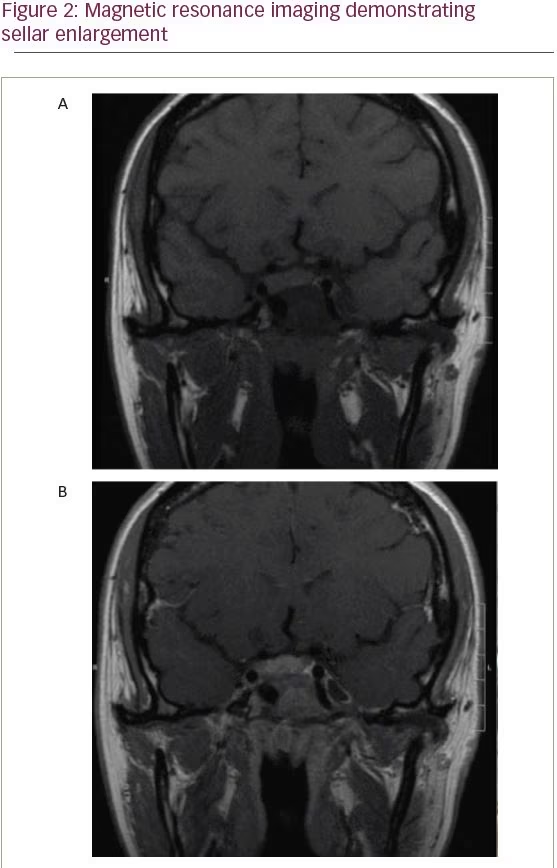
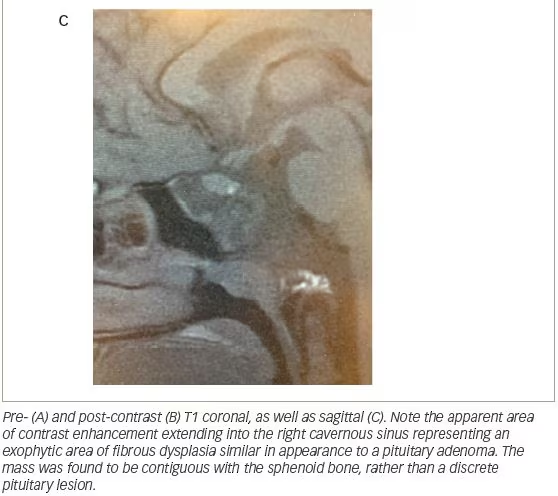
A 15-year-old girl with MAS and a history of fibrous dysplasia of the left femur, requiring open reduction and internal fixation, underwent computed tomography (CT) scan of the face and skull to evaluate a known history of fibrous dysplasia of the sphenoid bone. The pituitary gland was enlarged and it appeared there was a mass emanating from the undersurface of the gland. There was a large right-sided lesion that involved the body of the sphenoid, extended into the sphenoid sinus, and involved the right cavernous sinus. This lesion was thought to represent fibrous dysplasia of the sphenoid bone. A follow-up magnetic resonance image (MRI) was obtained (Figure 2).
She was referred to an endocrinologist for further evaluation of her abnormal pituitary gland. There were no historical features of acromegaly and there were no obvious gross acromegalic features on examination, including frontal bossing, prognathism, macroglossia, skin tags, hyperhidrosis, or hirsutism. There were no symptoms or signs of other anterior pituitary hormone excesses or deficiencies. Her height was similar to that of her older sister and was considered appropriate in relation to her estimated mid-parental height. There were several café-au-lait spots across her back. Laboratory investigations revealed a GH level that was elevated measuring 26.5 mcg/L. GH failed to suppress in response to oral glucose. The IGF-1 level was elevated at 791 ng/mL. The serum prolactin concentration was normal. Other pituitary functions were normal.
She was treated with Sandostatin® (octreotide, Novartis, Basel, Switzerland) long-acting release (LAR) 20 mg monthly. There were no side effects of treatment. There was no change in her overall sense of wellbeing. Her GH and IGF-1 levels fell to 16 mcg/L and 589 ng/mL respectively (age-adjusted normal range for IGF-1 was 217–589 ng/mL). The dose of Sandostatin LAR was increased to 30 mg monthly in attempt to maximize biochemical control of GH hypersecretion. Her GH and IGF-1 levels fell to 10 mcg/L and 585 ng/mL, respectively. Treatment was continued without side effects. Over the ensuing years, she experienced normal growth and achieved her estimated mid-parental stature.
Discussion
MAS is characterized by polyostotic fibrous dysplasia, café-au-lait hyperpigmentation of the skin, and hyperfunctioning endocrinopathies.8 The disorder is caused by sporadic mutations in GNAS, a gene that when mutated leads to mosaicism, wherein a mixed population of normal and abnormal cells are present to varying degrees in affected individuals. The clinical presentation is largely related to the extent and distribution of cells that possess the mutation.5,9 Hyperthyroidism, Cushing’s syndrome, and acromegaly are common hyperfunctioning endocrinopathies seen in MAS.6 In one large series of 112 treated patients, the mean age at presentation was 24.6 In general, gonadotropin-independent sexual precocity is the most common presentation, and affected patients commonly present with either symptoms of precocious puberty or those related to fibrous dysplasia as in our patient.6,10
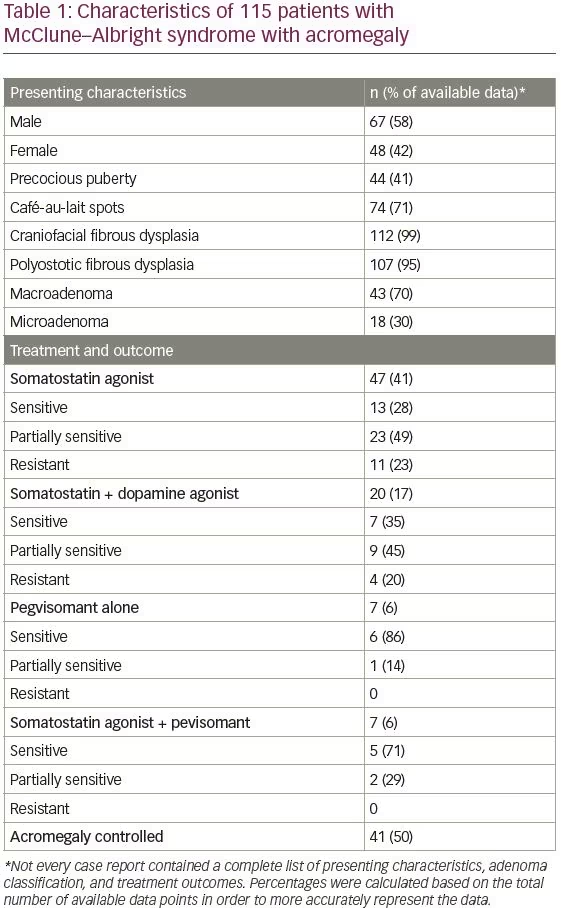
MAS-associated acromegaly has been reported previously in the literature, with varying degrees of treatment success (Table 1). In a study of 58 patients with MAS, 22 (38%) had clinical and biochemical stigmata of acromegaly.5 Twelve patients (21%) failed to suppress GH on an oral glucose tolerance test. Pituitary adenoma was identified on MRI of the sella in four of these
12 patients. Another study found clinical features of acromegaly in 20% of patients with MAS, with pituitary adenoma identified on MRI in 67% of those cases.11 This indicates a higher incidence of somatotroph adenomas in patients with MAS versus patients without MAS; the general incidence of GH-secreting adenomas in patients without MAS is approximately 10–15%.12 In addition, patients with MAS-associated acromegaly, present 10–20 years earlier than patients with classically described disease.6,13 Furthermore, the skeletal deformities that occur as a result of the fibrous dysplasia in MAS may mask the craniofacial changes often observed in acromegaly.6 Therefore, clinical testing for acromegaly, especially in a younger patient like the one presented in the current study, may be appropriate in all patients with MAS.
In the patient presented here, acromegaly was diagnosed as a result of the workup of an abnormal pituitary gland visualized on MRI. Serum prolactin levels were ultimately found to be within normal limits for our patient, though it is important to note that patients with MAS with GH hypersecretion can show elevated levels of prolactin, estimated as greater than 80% of patients.6 While the fibrous dysplastic lesions associated with MAS are best tracked by CT, MRI of the sella may be of clinical use in all patients with suspected MAS to identify pituitary abnormalities.14
Generally speaking, examination of resected pituitary tissue in patients with MAS who undergo surgery has illustrated widespread neoplasia within the gland. These findings suggest that resection of adenomas associated with MAS may not result in long-term GH control in these patients. Ablative therapy in affected patients must target the entire pituitary gland to increase the likelihood of successful control of the GH hypersecretion and the clinical manifestations of acromegaly.15 Furthermore, fibrous dysplasia of the sphenoid bone, as seen in our patient and others, may preclude successful transsphenoidal approach to the sella. For these reasons, most patients with MAS are not candidates for pituitary surgery.
In this patient, surgery was not considered a rational therapeutic option due to dysplastic changes precluding safe anatomical approach and resection, and because it appeared that the patient had a generalized disorder of her pituitary gland, rather than a discrete adenoma. For her, and many other patients with MAS-associated acromegaly, medical management of GH and IGF-1 excess is of paramount importance. Somatostatin analogs are effective in normalizing IGF-1 levels in about 50% of patients.7 However, in patients with MAS with acromegaly whose disease is not controlled by somatostatin analogs, pegvisomant, a GH receptor antagonist, may be useful as it has been shown to reduce IGF-1 levels in this population.3,4,10 Resistance to somatostatin analogs may be due to the G protein-coupled receptor-mediated pathogenesis of MAS, which may be better treated via direct receptor antagonism (Figure 3). Nevertheless, long-acting somatostatin analogs are still indicated as first-line therapy for acromegaly in patients with MAD due to the convenience of only requiring one injection per month. In this patient, Sandostatin LAR 30 mg monthly was sufficient to adequately control disease.
The efficacy of radiotherapy in these patients is uncertain and there remain concerns regarding the possibility of sarcomatous transformation of fibrous dysplasia.6,12 Thus, it is not our policy to offer radiotherapy to children or young adults with MAS unless surgery is contraindicated and there has been no demonstrable biochemical response to pharmacologic agents.
In summary, we offer further evidence that acromegaly associated with MAS may present earlier and with different clinical signs and symptoms than classic stigmata of acromegaly, such as frontal bossing, hyperhidrosis, or hirsutism; and instead exhibit unique features such as craniofacial deformity and endocrinopathy including hyperprolactinemia. MRI of the sella may be indicated in all patients with MAS to assess for the existence of a pituitary adenoma. Medical management is often preferable over surgery in this patient population due to fibrous dysplasia of the bones preventing safe and reliable access to the sella. If patients are refractory to treatment with somatostatin analogs, the GH receptor antagonist pegvisomant may be effective in achieving disease control.