Congenital adrenal hyperplasia is an innate error in the biosynthesis of adrenal steroids, which triggers a wide range of consequences based on the level of the enzyme blockade. A deficiency in the enzyme 21-α-hydroxylase is the most frequent cause, presenting with different degrees of virilization in women, and may or may not present with the salt-wasting form. In contrast, 17-α-hydroxylase deficiency (17OHD) is an uncommon form of the disease, which presents contrasting phenotypic characteristics in relation to the other forms.1
Cytochrome p450c17 (CYP17A1) catalyses two pathways of steroidogenesis: 17-α-hydroxylation, which is involved in cortisol production, and 17.20-lyase, involved in androgen production in both adrenal glands and gonads. The resulting cortisol deficiency determines increased release of adrenocorticotropic hormone (ACTH), resulting in accumulation of steroid precursors, such as progesterone, deoxycorticosterone (DOC) and corticosterone.1 This article reports on a case of late diagnosis of the congenital adrenal hyperplasia. Informed consent was obtained from the patient.
Case description
In April 2015, a 19-year-old female patient was referred for ambulatory attendance to the gynaecology service due to primary amenorrhea and delayed pubertal development. Physical examination showed absence of pubic hair and infantile breast and genitals. A pelvic ultrasound was performed, but the uterus or ovaries could not be characterized. A karyotype test demonstrated 46,XY cells (34 metaphases) in addition to undetectable serum oestradiol. This was interpreted as complete androgen insensitivity syndrome, and magnetic resonance imaging of the pelvis for characterization of the gonads was performed, identifying only the left gonad. The patient underwent left orchiectomy due to malignization risk, and was started on conjugated oestrogens 0.625 mg/day, and followed up by the gynaecology service.
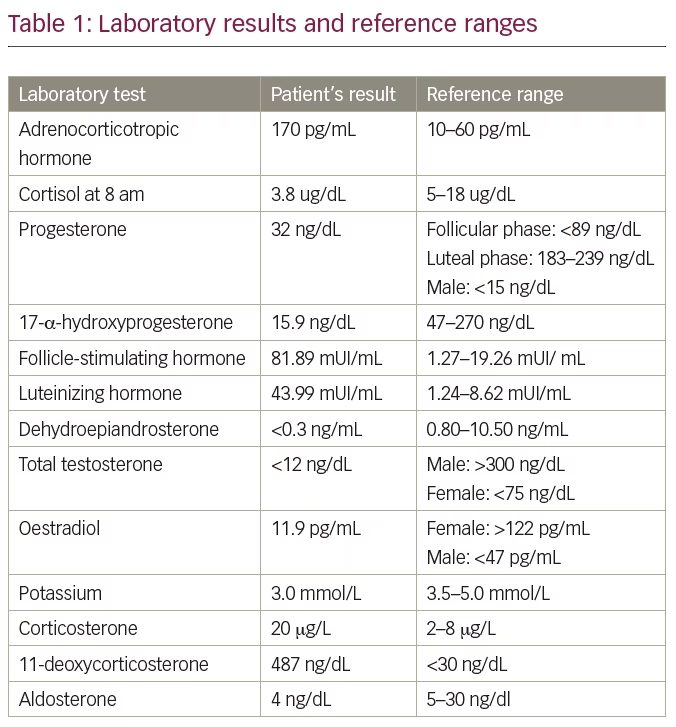
Three years later, when she was 22 years old, the patient was referred to the endocrinology clinic to continue treatment. Thorough physical examination revealed a stature of 191 cm (above the target height 165 ± 5 cm), arm span of 197 cm, weight of 75.3 kg, blood pressure of 160/110 mmHg, underdeveloped breast tissue, as well as absence of pubic or axillary hair and external genitalia characteristic of a female child. Breast ultrasound showed breast parenchyma with preserved echotexture and retroareolar, and wrist x-ray showed bone age compatible with that of a 14-year-old female. Laboratory tests were performed for further evaluation of a 46,XY sexual development disorder, and are depicted in Table 1 with the respective reference range. Subsequent to these results, we biochemically confirmed the hypothesis of 17OHD.
The patient was then started on treatment with glucocorticoid replacement (prednisone 5 mg) and maintenance of oestrogen replacement with progression of dosage (3.75 mg of conjugated oestrogen/day). She was also referred to the psychology department. This led to a reduction in blood pressure; however, it was necessary to initiate spironolactone 25 mg for complete control, as well as breast development.
Discussion
The most common cause of congenital adrenal hyperplasia, globally, is deficiency of the enzyme 21-α-hydroxylase.1 Other less frequent causes vary in prevalence in different populations; for example, in Brazil, there is a high prevalence of 17OHD, being considered the second most common form of the disease in the country.2 The actual incidence of 17OHD is estimated to be 1/50,000–70,000,1 corresponding to approximately 1% of all congenital adrenal hyperplasia cases.2–4 Genetic determinants of disease mutations appear to be related to ethnicity of the population studied. In Brazil, the incidence of this disorder, although still small, is significantly higher than in other regions of the world, due to genetic mutations inherited from the country’s colonizers, which persist despite the immense racial miscegenation, differently from what is expected in diseases with a autosomal recessive inheritance, like 17OHD.1,3 Approximately 100 types of mutations with functional impairment of 17OH and 17.20-lyase have already been described.3 The Brazilian Multicenter Study Group for Congenital Adrenal Hyperplasia assessed 24 carriers of the disease – the largest report of a single country – and described seven new mutations, in addition to the approximately 40 already described.2
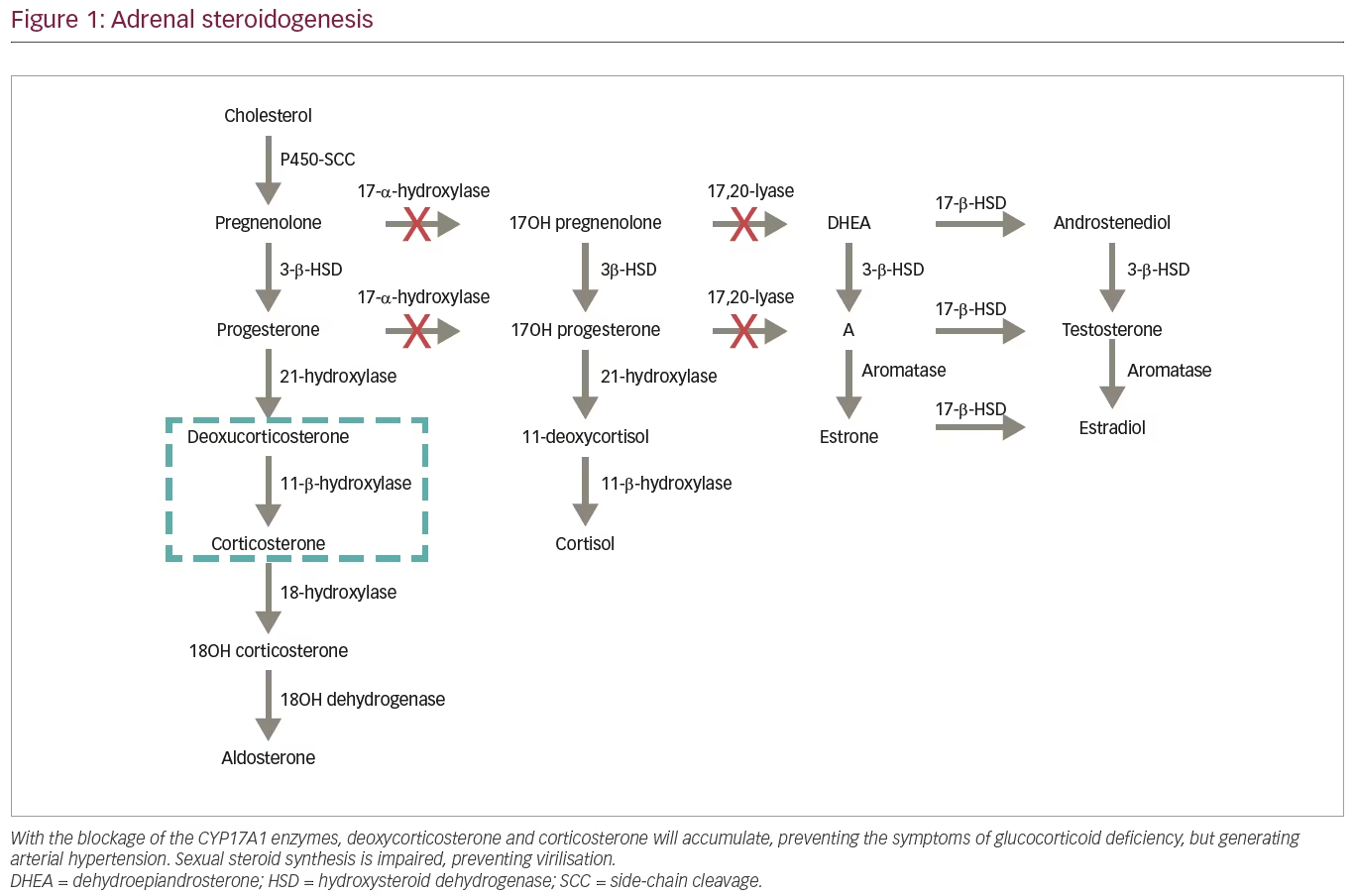
Most steroidogenic enzymes are members of the cytochrome P450 oxidase group; these enzymes are characterized by containing about 500 amino acids and a single haem group, in addition to absorbing light at 450 nm in their reduced states.5 The 17OHD arises from mutations in CYP17A1, located on the chromosome 10p 24.3, which encodes the enzyme that catalyses both reactions promoted by 17OH and 17.20-lyase.1–3 In the path of steroidogenesis from cholesterol (Figure 1), blocking the 17OH enzyme generates accumulation of 17-deoxysteroids, 11-DOC and corticosterone, preventing formation of other steroids, such as cortisol and androgens.3 It is important to note that severe mutations with complete 17OHD also disrupt steroidogenesis in the gonads, as production of both androgens and oestrogens requires the 17,20-lyase activity of CYP17A1.
The absence of exposure to intra-uterus testosterone determines a female phenotype in most cases, regardless of the genotype.1 XX genotypes present with normal female genitalia at birth, but fail to reach pubertal development, and amenorrhea frequently occurs. On the other hand, XY individuals are usually raised as girls, featuring female external genitalia, with blind short-ended vagina, and absence of female internal genitalia, caused by the action of Mullerian inhibitor factor produced by the testis. Men with partial 17OHD may display ambiguous external genitalia or pseudohermaphroditism due to partial genitalia virilisation.1,5 Often, the affected individuals, in both genotypes, feature high stature with eunuchoid habitus, resulting from the delayed closure of bone epiphyses by oestrogen deficiency.1,6
The classic clinical manifestations of mineralocorticoid deficiency are not displayed in patients with 17OHD. Due to the loss of cortisol’s negative feedback on ACTH, there is an important increase in the production of all hormones produced prior to the deficient enzyme.4 The elevation of serum DOC binds to the mineralocorticoid receptor, promoting sodium retention, elimination of potassium and increased intravascular volume, producing a state of hypertension with hypokalaemia and hypoaldosteronism (due to a negative feedback by DOC), accompanied by suppression of plasmatic renin.3,4 Excess corticosterone acts on glucocorticoids, preventing the clinical manifestations associated with deficiency of cortisol, such as hypoglycaemia.1,4,5
Atypical manifestations of 17OHD include ovarian cysts in 46,XX patients, which occur due to chronically elevated gonadotropins.7,8 The diagnosis of 17OHD is established by high serum progesterone, pregnenolone, DOC and corticosterone levels, as well as increased urinary excretion of its metabolites. Despite the significant increase in levels of corticosterone, which occurs almost exclusively in 17OHD, measurement of this plasmatic precursor is rarely performed, which may explain the large number of erroneous diagnoses of up to 60%, as demonstrated in previous studies.1,4 Diagnosis is made in the presence of low cortisol, dehydroepiandrosterone (DHEA) and 17OH-progesterone (17OHP), and high DOC and corticosterone; in partial forms, ACTH stimulation testing may be useful to show adrenal steroidogenic defect.9 It is important to notice that the other hypertensive form of congenital adrenal hyperplasia, 11-hydroxylase deficiency (11OHD), also present with elevated DOC but with high 11-deoxycortisol, which is low in 17OHD; also, androgens are elevated in 11OHD, but low in 17OHD.9
The treatment of congenital adrenal hyperplasia consists of glucocorticoid replacement in physiological doses, as well as sexual steroid replacement.5 The use of glucocorticoids aims to provide sufficient effect to exert negative feedback on ACTH, and consequently regulate blood pressure and serum potassium; however, the use of aldosterone-receptor blockers may be necessary for further blood pressure control.5 Most patients with 17OHD grow as individuals of the female gender; and therefore, should receive supplementation of oestrogens followed by supplementation of progestogen (in the case of genotype 46,XX), resulting in epiphyseal closure, as well as in the development of secondary sex characters. Testosterone supplementation in low doses can also be useful for the development of pubic hair.1 The use of DHEA has also been studied in some cases of primary adrenal insufficiency; studies using doses of 25–50 mg/day show beneficial results, such as libido and other biochemical parameters; its use, however, has not been studied specifically in 17OHD.10 Both genders do not progress through puberty and are generally infertile.3
A risk that must be assessed is gonadal tumorigenesis; germ-cell tumours have a higher incidence in cases of gonadal dysgenesis (15–60%), compared with defects in synthesis or androgenic action (around 5%), with the presence of Y chromosome material being the greatest risk factor, due to the presence of the TSPY gene (testis–specific protein in chromosome Y), which promotes gonadoblastoma.11 Currently, the most accepted approach is bilateral gonadectomy in patients with the Y chromosome or SRY portion and dystopic gonads.
Conclusion
When facing a case of amenorrhea associated with pubertal delay, a range of diagnostic hypotheses must be considered; therefore, a detailed clinical history and physical examination are important for effective investigative targeting. This report demonstrates the importance of this accurate clinical evaluation in all medical consultations; due to non-routine blood pressure measurement in paediatrics, the disease was only evidenced from pubertal delay. Another crucial point in the clinical evaluation of this patient was the eunuch habitus and enlargement of the arm span, which could not fail to be noticed. Serious physical and psychological consequences due to unconformity of fenotype with genotype, as well as infertility, may occur in cases of delayed diagnosis and undertreatment.