Moyamoya is a rare idiopathic cerebral vasculopathy characterized by stenosis of the terminal portion of the internal carotid arteries (ICAs) and the development of a thin collateral network of small vessels (“puff of smoke”), initially described as a primary disease in young individuals of Asian descent.1
Graves’ disease (GD) is an autoimmune hyperthyroid state in which antibodies stimulating the thyroid simulating hormone (TSH) receptor, thyroid peroxidase, and thyroglobulin exist, causing hyperthyroidism. A vasculopathy similar to Moyamoya has been identified in association with GD, predominately in Asian population (<70 cases so far). “Moyamoya-like vasculopathy” is a proposed term for this “secondary” vasculopathy.2
Here we describe first familial presentation of two patients of Latino descent, mother and daughter, with GD and intracranial occlusive arteriopathy.
Diagnosis of Graves’ Disease
Traditionally GD has been characterized by hyperthyroidism, diffuse goiter, ophthalmopathy, and dermatopathy. Hyperthyroidism is usually presented with clinical features of weight loss, anxiety, and tremors along with lab testing showing low TSH and high free tri-iodothyronine (T3) and tetra-iodothyronine (T4) levels. Additional testing with antibodies to thyroglobulin, thyroid peroxidase, and TSH receptor help in making the diagnosis; however, their presence is not mandatory for diagnosing GD.3,4
Both of our patients presented with undetectable/low TSH and elevated T4 at the time of stroke. Antibodies to thyroid peroxidase, TSH receptor, and thyroglobulin were positive in both of our patients. Patient 1 was diagnosed at a very early age (mentioned below), where as in Patient 2, the diagnosis was made later on when she presented to hospital during the acute episode.
Patient 1
Initial Presentation
A 19-year-old Hispanic woman presented to the hospital in June 2008 with left-sided weakness, which had worsened over 1 week. At age of 16, she was found to have hyperthyroidism, ophthalmopathy, low TSH, high T3 along with positive thyroglobulin, and TSH receptor and thyroid peroxidase antibodies, hence diagnosis of GD was made. She was intermittently taking propylthyouralcil (PTU) and propranolol. There was no history of cardiac diseases, diabetes, dyslipidemia, or smoking. On admission, blood pressure (BP) was 159/85 mmHg, heart rate 90/minute, and temperature 98°F. Electrocardiogram (ECG) and echocardiogram were unremarkable. On examination, there was a left hemiparesis (3/5 strength in arm and leg) with hyperreflexia and Babinski sign. Sensation to light touch and pinprick was decreased on the left, with extinction to doublesimultaneous stimulation, but no neglect. TSH level was <0.002 mIU/l (0.2–4.0 mIU/l) and T4 was 18.9 μg/dl (4.6–12.0 μg/dl). Blood cell count, erythrocyte sedimentation rate (ESR), antinuclear antibody (ANA),
anticardiolipin antibodies (ACL), rapid plasma reagin (RPR), rheumatoid factor, and hemoglobin electrophoresis (HbEP) were unrevealing. Her rest of work up for stroke in young patient, which included testing for hypercoaguable state, prolonged Holter monitor for detection of atrial fibrillation, and transesophageal echocardiogram for any cardiac source of embolism, were negative.
Radiological Findings
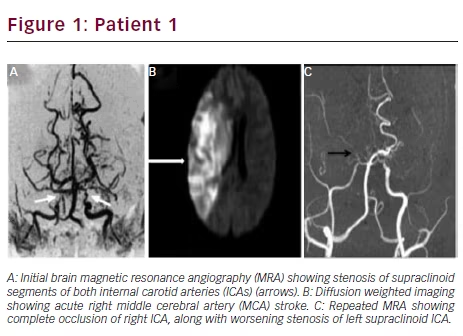
Initial brain magnetic resonance imaging (MRI) revealed a small right middle cerebral artery (MCA) ischemic stroke. Magnetic resonance angiography (MRA) showed stenosis of the supraclinoid segments of both ICAs, consistent with Moyamoya pattern (see Figure 1A). She was treated with aspirin, PTU, propranolol (80 mg/day), and intravenous (IV) fluids.
Clinical Course and Prognosis
Two days later, she became lethargic, with left hemiplegia and neglect. Second brain MRI showed progression of the right MCA ischemic stroke, now involving the complete territory of the artery (see Figure 1B). Occlusion of the right ICA and severe stenosis of the left supraclinoid ICA were seen in follow-up MRA (see Figure 1C). She rapidly developed malignant brain edema and uncal herniation. Emergent decompressive hemicraniectomy was lifesaving, but she remained in a persistent vegetative state.
Patient 2
Initial Presentation
A 47-year-old woman, mother of Patient 1, had no significant medical history until November of 2012, when she developed slurred speech and right facial droop. Speech difficulty resolved spontaneously in 15 minutes but facial asymmetry persisted. There was no associated limb weakness or numbness, no visual complaints, headaches, chest pain, or palpitations. She occasionally smoked cigarettes but denied any history of alcohol or drug abuse. On admission she was noted to be afebrile, and BP was 124/81 mmHg with regular pulse. ECG and echocardiogram were unremarkable. On examination, there was a right central facial palsy, but language function, motor strength, and sensory examination and coordination were normal. Cardiopulmonary examination was unremarkable and there were no carotid bruits. Laboratory analysis showed mild ferropenic anemia, normal white-blood-cell count, and platelets. Comprehensive metabolic profile and HbEP were normal. ESR was 38 mm in the first hour. ANA, anti-double stranded DNA antibodies, RPR, ACL, protein C and S, antithrombin III activity, and lupus anticoagulant were unrevealing. Total cholesterol was 142 mg/dl and low density lipoprotein 77 mg/dl. TSH was 0.02 (0.35–5.60 mIU/l), free T3 was 5.9 (2.3–4.2 pg/ml), and free T4 was 1.64 (0.58–1.64 ng/dl). Further testing revealed positive antibodies to thyroid peroxidase, TSH receptor, and thyroglobulin, thus confirming clinical suspicion (clinical symptoms mentioned above) of GD.
Radiologic Findings
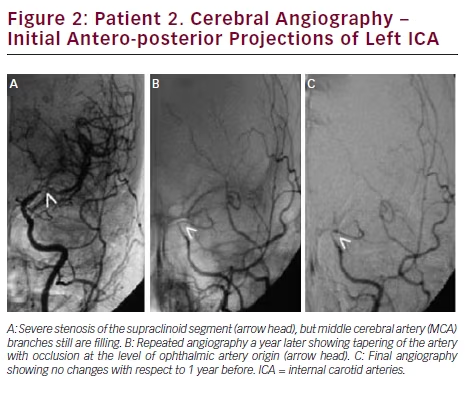
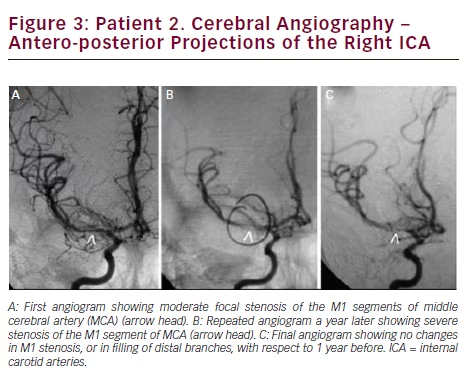
Brain MRI showed small acute ischemic infarcts at the left fronto-parietal convexity. Brain MRA revealed stenosis of the terminal portions of both ICAs and proximal segments of both MCAs. Carotid ultrasound showed no significant stenosis bilateral. A cerebral angiogram revealed severe stenosis of the distal cavernous and supraclinoid left ICA and proximal segments of left MCA (see Figure 2A). On the right, there was moderate stenosis of the M1 segment of MCA (see Figure 3A). She recovered after 4 days of hospitalization and was discharged on treatment with aspirin only.
Long-term Follow Up and Prognosis
In September of 2013, she was readmitted after developing slurred speech that lasted 15 minutes. This time, her neurologic examination was completely normal. She complained of palpitations, shortness of breath, profuse sweating, fine hand tremors, and a 30-pound weight loss in the previous 6 months. The patient reported that she was noncompliant to medications since the time she was discharged. A small acute ischemic lesion in the left fronto-parietal area was seen on brain MRI, and MRA showed progression of the intracranial stenosis. Cerebral angiogram revealed a narrowed segment at the cervical portion of left ICA, with occlusion at the cavernous portion (see Figure 2B). On the right side, there was a severe focal stenosis of the M1 segment of MCA (see Figure 3B). Her TSH was 0.006 (0.34–5.60 mIU/l), T4: 20.91 (6.09–12.23 μg/dl), and T3: 502 (80–200 ng/dl). She was treated with aspirin, propranolol, and methimazole and symptoms of hyperthyroidism resolved.
During the following 12 months, she remained compliant with medication, with no new symptoms and readmissions. In September 2014, her thyroid function tests were within normal limits. Repeat brain MRI showed no ischemic lesions. No further progression of the intracranial stenosis was seen in a follow-up cerebral angiogram (see Figures 2C and 3C).
Discussion
The association of GD and intracranial occlusive arteriopathy, complicated with ischemic strokes, is increasingly being reported in the scientific literature.5–7 The pathogenesis has not been elucidated yet, but the association does not seem to be coincidental. The genetic association in GD is more well established than the genetic association in Moyamoya disease; it can be argued that familial component may be GD, which causes T-cell dysregulation along with enhancement of sympathetic nerve stimulation at the level of the carotid arteries, leading to vascular injury resulting in Moyamoya syndrome.8,9 On the other hand, the majority of patients diagnosed with Moyamoya disease have been women. A possible genetic association is shown in Moyamoya disease from studies carried out in Japan and the US. A familial variant with 17q25 chromosomal defect has been associated with Moyamoya disease. These data suggest a possible sex-linked predisposition along with possible genetic predisposition;10,11 however, further studies are needed to clarify whether genetic predispositions underlie the association between GD and Moyamoya vasculopathy.
Another important point worth considering in subjects with emboliclooking strokes and history of hyperthyroidism is that in these patients, work up for cardiac source of embolic stroke (through prolonged Holter monitor or event monitor) should be performed, given high concurrence of cardiac arrhythmias, especially atrial fibrillation with hyperthyroidism.12 After thorough investigation and ruling out common causes of embolic strokes, diagnosis of Moyamoya should be considered.
We described two patients with GD who developed progressive intracranial arterial stenosis and ischemic strokes. Both patients were thyrotoxic at the moment of the ischemic events. In both patients, other common causes of embolic strokes were ruled out. On the first reported case, the course of the disease was rapidly progressive, leading to ICA occlusion and large hemispheric stroke over only a few days. On the second case, there was evidence of progression of the intracranial stenosis, also leading to occlusion of one ICA over 1 year, when she was thyrotoxic. The disease process stabilized once the hyperthyroidism was well controlled and follow-up studies after 1 year did not show further progression of the stenosis. Hence one can hypothesize management of hyperthyroidism could to be an important factor for the progression of the disease. To the best of our knowledge, this is the first report of familial presentation of the association of GD with intracranial occlusive arteriopathy and ischemic strokes in patients of Latino descent.