Acromegaly is a chronic disorder characterised by growth hormone (GH) hypersecretion, predominantly caused by a pituitary adenoma.1 Disease prevalence ranges from 2.8–13.7 cases and annual incidence is between 0.2–1.1 cases/100,000 people; however, real incidence is likely much higher.2,3 Average age at diagnosis ranges from 40–50 years4–6 and mean delay in diagnosis is approximately 10–11 years. More than 95% of acromegalic cases are secondary to a pituitary adenoma: somatotrophs or GH-producing cells. In <5% of cases, acromegaly is related to a hypothalamic or neuroendocrine tumours, which secrete GH-releasing hormone, leading to excess GH. Peripheral GH-secreting tumours are exceedingly rare.1
GH stimulates synthesis of insulin-like growth factor 1 (IGF-1) from the liver and systemic tissues. Hypersecretion of GH leads to excess production of IGF-1. IGF-1 mediates most of the phenotypic features and metabolic effects of GH, but GH excess also has direct detrimental effects.1,7 Acromegaly is associated with increased morbidity and mortality, but mortality returns to that of the normal population after appropriate treatment and biochemical normalisation.8,9 This review focusses on several recent updates related to acromegaly diagnosis and treatment.
Screening and diagnosis
Screening is recommended for all patients presenting with clinical features of acromegaly (such as mass tumour effects, systemic effects of GH/IGF-1 excess, cardiovascular and metabolic features, respiratory and bone/joint manifestations and/or other endocrine consequences). However, screening may also be considered in patients with several medical conditions known to be associated with acromegaly such as type 2 diabetes mellitus, carpal tunnel syndrome, debilitating arthritis, hypertension and sleep apnoea.10–12 Awareness of these comorbidities is critical for early detection of acromegaly.
Biochemical screening is the first step for an acromegaly diagnosis. Endocrine Society guidelines and experts’ consensus recommend using age- and sex-adjusted IGF-1 levels in combination with GH nadir during an oral glucose tolerance test (OGTT) to diagnose and rule out acromegaly.13,14 Measuring serum IGF-1 is usually the initial screening test. Considerable variation in laboratory results for IGF-1 obtained from different assays,15 pose a hindrance to diagnosis. For example, these discrepancies may lead to inaccurate exclusion of a diagnosis. This has been reported in up to 30% of patients in different laboratories.16 Given the methodological differences between assays and to establish accurate laboratory results, interpretation reference intervals must be method-specific, adjusted for age and sex, and stratified according to Tanner stages.17 Equivocal or elevated IGF-1 levels require further diagnosis confirmation in most patients. An OGTT with 75 g glucose is considered the gold standard for diagnosing acromegaly. However, similar to IGF-1 assays, the GH assay method can impact the absolute GH concentration reported by a laboratory.18 As a consequence, the assay method may also impact the cut-off for GH suppression following oral glucose load.19 Current widely used cut-offs for GH after OGTT are 1.0 and 0.4 ng/dL. However, these may not be accurate for all commercial assays, and method-specific values for GH cut-offs must be reported when available.13,20
Severe obesity, prolonged fasting and malnutrition reduce IGF-1 levels in patients without acromegaly21,22 and may also impact levels in patients with acromegaly. Random GH level testing is not recommended for diagnosis given the pulsatile nature of secretion.23 Stress, physical exercise, acute critical illness and fasting state can cause physiological higher peak in GH secretion.24–26 In pregnancy, homology between GH and placental GH makes GH measurement especially challenging in acromegaly cases.27 Chronic renal failure can lead to higher GH but IGF-1 remains unchanged or can even decrease.28 Type 2 diabetes and insulin resistance are associated with higher GH due to impaired suppression by glucose, while chronic hyperglycaemia has shown to be associated with decreased GH release.29 High GH with low IGF-1 can be observed in states of GH resistance such as systemic inflammation, chronic liver disease, cirrhosis and anorexia nervosa.30–32
Biochemical markers, IGF-1 and GH results may be discordant due to their biological and analytical variability, as mentioned above. Patients with clinically active acromegaly and elevated IGF-1 may still have ‘suppressible’ GH after OGTT using both cut-offs of 1 and 0.4 ng/dL. These discordant findings were observed in 18–45% of treatment-naïve patients with acromegaly,33,34 and in 17–35% of patients with acromegaly after treatment (surgery, radiotherapy and/or medication).35–37 Recent studies showed that a large number of patients with acromegaly can have a very different clinical, biochemical and radiological presentation compared with what is considered a ‘classical’ one. Patients with a typical clinical phenotype and high IGF-1 levels, can have plasma GH in the ‘normal’ range with glucose-suppressed GH <1 µg/L in ~50% and <0.4 µg/L in as many as 30% of patients.33,34
In summary, the most important update in screening and diagnosis is an increased knowledge and acceptance of the fact that classical diagnostic criteria of acromegaly no longer apply to all patients.
Imaging
After GH hypersecretion has been confirmed, the next step is determining the source of excess GH. Pituitary magnetic resonance imaging (MRI) is recommended, given >95% of acromegaly cases are caused by a pituitary adenoma.1 A computed tomography scan can be obtained when MRI is contraindicated or unavailable.13
Certain radiologic characteristics have been recently recognised as markers for disease behaviour and predictors of response to therapy. Adenomas that have a hypointense signal on T2-weighted MRI were found in >50% of GH-secreting tumours, to be smaller and less frequently invasive, although associated with higher levels of GH hypersecretion.38 IGF-1 decreased by more than half from pre-treatment levels in 66% of T-2 hypointense versus 31% of T2-hyperintense adenomas after 6 months of adjunctive somatostatin receptor ligand (SRL) therapy, and normalised in 71% versus 20%, at 6 months, respectively.39
Patients with T2-hypointense adenomas had GH and IGF-1 reductions of 88% and 59%, respectively after 6 months of pre-surgical SRL treatment, significantly greater than the decreased levels observed with T2-isointense and hyperintense tumours. Tumour shrinkage was also greater in T2-hypointense tumours. Lower T2-signal intensity was found to correlate with a better hormonal response, but the correlation with tumour shrinkage was inconsistent among studies.40–42
Pathology
In 2017, the World Health Organization updated the histological grading of pituitary neuroendocrine tumours.43 The new grading abandoned the term ‘atypical adenoma’ and emphasised the evaluation of morphology, tumour proliferation and invasion status for prognostication and evaluation of aggressiveness.44
GH-producing pituitary adenomas have several histological subtypes, and differ in morphology, clinical and biological behaviour. Classification is derived from the results of hematoxylin-eosin stain, immunohistochemistry, appearance under an electron microscope and transcription factors expressed in cells, and the following subtypes are established:
-
- GH-producing adenomas:
densely granulated somatotroph adenomas (DGSA)
sparsely granulated somatotroph adenomas (SGSA); and
intermediate granulated somatotroph adenomas.
-
- mixed GH/prolactin producing adenomas:
mammosomatotroph adenomas; and
acidophil stem cell adenoma.
- plurihormonal adenomas, and silent somatotroph adenomas.45
DGSA are found in 40% of acromegaly tumours, while SGSA are found in 30%.46,47 SGSA are usually larger at diagnosis than DGSA (21.6 mm versus 19.2 mm, respectively), have lower somatostatin receptor subtype 2 positivity (50% versus 100%) and higher Ki-67 proliferation index (>3% in 67% of SGSAs versus <3% in 89% in DGSA).48 Sparse granulation pattern has been also correlated with adenoma hyperintensity signal on T2-weighted MRI.49
Patients with SGSA usually require more surgeries, more radiotherapy, multiple different medications, a higher number of combined treatments and show medication resistance more often than DGSA. In a recent large study, median time for biochemically controlled acromegaly, using age-adjusted IGF-1 levels, was 9.7 years versus 16 years, in SGSA and DGSA, respectively.50
Intermediate granulated somatotroph adenomas have similar clinical behaviour to DGSA. Both acidophil stem cell and plurihormonal adenomas have been associated with aggressive behaviour, while clinical behaviour of silent somatotroph adenomas is variable, but often aggressive.51–53 It is essential that pathology defines the exact type of GH pituitary adenoma in all patients undergoing surgery, as it has been proven to predict both clinical and biochemical outcomes.
Treatment
Transsphenoidal pituitary adenoma resection is generally the first-line therapy. Successful surgery provides immediate reduction of GH levels and provides tumour tissue for diagnostic and prognostic purposes.13 However, not all patients achieve remission after surgery and acromegaly treatment is frequently multimodal.54
When operated on by an experienced pituitary surgeon, the outcome of transsphenoidal surgery for acromegaly is similar to endoscopic and microscopic techniques. Highly experienced pituitary centres, lower preoperative GH level, small size of tumour, and extrapseudocapsular resection are factors associated with higher remission rates post-operatively, while lower remission rate is seen with macroadenomas and tumours invading the carvernous sinus and parasellar area. GH values <1 ng/mL within the first 72 hours after surgery is a positive predictive factor for remission.54–56
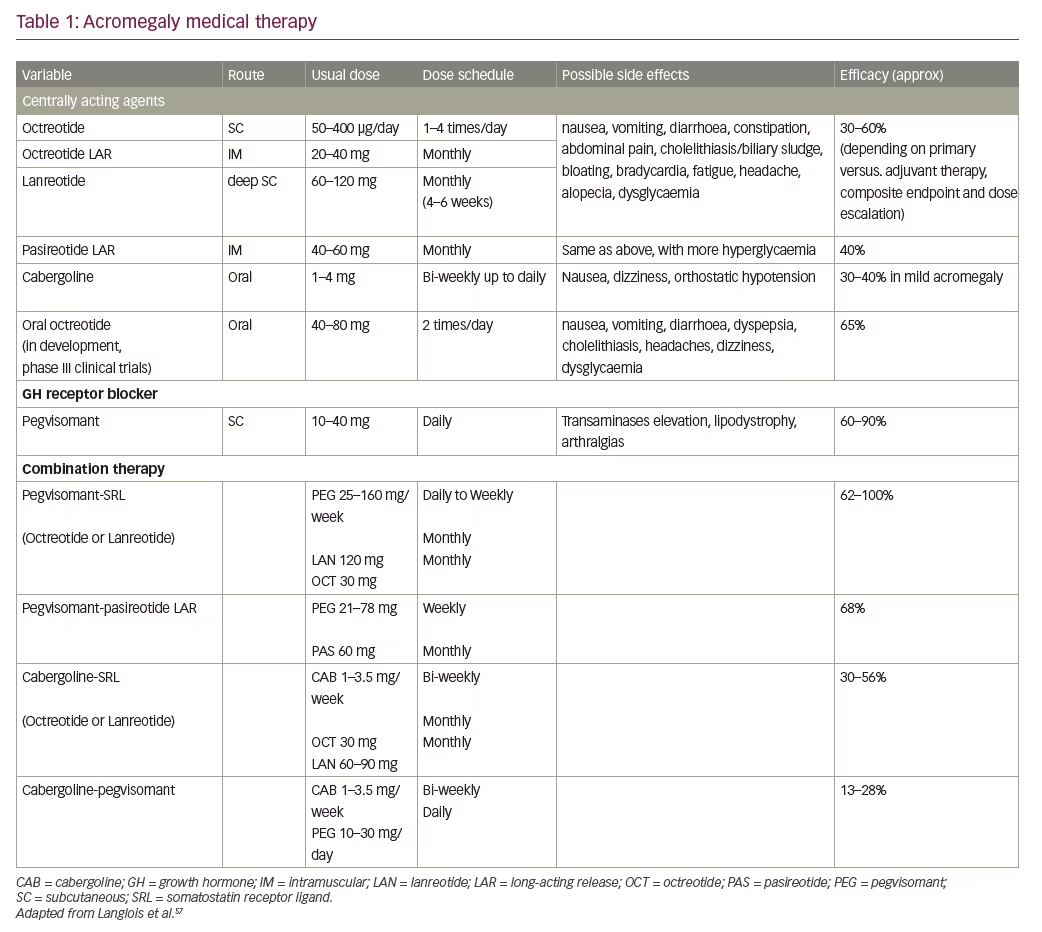
Pharmacologic agents available for treatment of acromegaly include somatostatin-receptor ligands, GH-receptor antagonists and, in selected cases, dopamine agonists (Table 1).57 The first-generation of SRLs, octreotide and lanreotide, have been the mainstay of medical treatment,1 however, most recently pegvisomant (a genetically engineered, recombinant GH-receptor antagonist) has also been used as a first-line treatment.58,59 Biochemical response and tumour reduction with SRLs varies widely between studies, from 20–70%, depending on the study design, history of surgical debulking and endpoint of the study itself (IGF-1, GH or composite GH and IGF-1). In a recent meta-analysis evaluating the effect of study design on the reported biochemical response rates to SRL, overall efficacy response rate was 56% for GH control and 55% for IGF-1 normalisation, without significant difference between SRL types.60 However, efficacy was only approximately 40% for GH normalisation in treatment-naïve, newly diagnosed patients treated with lanreotide autogel.61,62 Tumour volume reduction of >20% was found in 75% and 54.1% of patients treated with octreotide long-acting release (LAR) and lanreotide autogel, respectively, but with different duration of follow-up.62–64
Symptoms and quality of life (QoL) were recently evaluated in a 1-year, open-label study of lanreotide autogel in acromegaly treatment-naïve patients. Symptoms improved in 60% using a Patient-assessed Acromegaly Symptom Questionnaire, while 40% had improved health-related QoL using the AcroQoL questionnaire. Clinical symptoms improved in both patients with and without biochemically controlled disease.65 However, a cross-sectional study comparing QoL between patients with treated and controlled acromegaly and healthy controls, and a longitudinal study assessing QoL changes in patients with biochemically stable disease during approximately 6 years of follow-up demonstrated that impaired QoL in patients with acromegaly persisted despite long-term disease control. Notably, duration of disease control and present use of medical therapy for acromegaly influence QoL the most.66
In patients resistant to first-generation SRLs, alternative options are high-dose regimens of SRL, pegvisomant, combination therapy of SRL with either cabergoline or pegvisomant, or the use of pasireotide.67 Pasireotide is a next-generation SRL recently approved for the treatment of acromegaly not cured by surgery or when surgery is not an option. As a first-line therapy, pasireotide LAR resulted in higher rates of hormonal control and tumour size reduction compared to octreotide LAR.68 In 130 patients resistant to first-generation SRL randomised to pasireotide LAR 40 mg or 60 mg, a complete biochemical response was achieved in 15% and 20%, respectively.69 The safety profile is similar to first-generation SRLs, except for hyperglycaemia, which occurred more frequently and more severely with pasireotide.68,70 The frequency of hyperglycaemia was similar in responders and non-responders to pasireotide. Baseline glucose status (fasting levels >100 mg/dL) is a potential predictive factor to higher glucose and haemoglobin A1C after treatment.71 The reduction in insulin and incretin hormones secretion are thought to be contributors, but the exact mechanisms are not completely understood72,73 and further studies are ongoing.
Pegvisomant is a US Food and Drug Administration-approved treatment for use after surgery. In a global non-interventional safety surveillance study, pegvistomant normalised IGF-1 in 67.5% of patients after 5 years (most likely due to lack of dose-up titration), and also improved clinical symptoms.58,59 Pegvisomant improves insulin sensitivity, and long-term follow-up showed significantly decreased fasting glucose over time, while first-generation SRL only have a marginal clinical impact on glucose homeostasis in acromegaly.74,75 Pegvisomant does not have any direct anti-proliferative effects on pituitary tumour cells, but tumour growth is rare overall. In clinical trials of patients treated with pegvisomant and with available MRI the incidence of increase in pituitary tumor size was 3.2%.58,59
Cabergoline has been recommended by guidelines and consensus meetings to be considered in patients uncontrolled on SRL monotherapy who have baseline IGF-1 levels up to 1.5–2.2 times above the upper limit of normal.13,14 Adding cabergoline to ongoing SRL therapy has been reported to achieve IGF-1 normalisation in 30–56% of patients who had uncontrolled acromegaly on SRL-monotherapy.67,76,77
Patients with a higher degree of resistance to SRL monotherapy should be considered for combined SRL-pegvisomant13,14 or pasireotide treatment.67 Combined therapy of SRL and pegvisomant in patients with uncontrolled disease on SRL had a high rate of efficacy in different studies, 62–100%, in both primary and adjuvant therapy.77,78 Differences in reported outcomes are likely related to many factors including heterogeneity in study designs such as IGF-1 assays, inclusion/exclusion criteria, and inconsistent medications dosing and titration protocols. Another factor is the different IGF-1 criteria used to define efficacy, with some studies using the lowest achieved IGF-1 level at any point versus the level at the end of study in others. Switching 61 patients with well-controlled disease on a combined therapy of pegvisomant-first-generation SRL to pasireotide LAR resulted in a 66% reduction in pegvisomant dose, while 67% of patients could discontinue pegvisomant at 24 weeks.79 All combination treatment therapies have potentially higher rates of adverse events than single treatment therapy, and therefore patients require close monitoring.
There are several new therapies in development, including new delivery systems,80 or extended interval (lanreotide prolonged release formulation).81 Oral octreotide, an oral therapeutic peptide has shown efficacy in a large phase III trial in controlling IGF-1 and GH after switching from injectable SRLs in 65% of patients for up to 13 months, with a safety profile consistent with other SRLs.82 Primary surgical debulking has been shown in a recent study to be superior to primary medical therapy for macroadenoma, with a 50% and 6.7% response rate, respectively. The response rate remains highest when medical therapy is added after surgical failure at 77%.83
The impact of pre-operative SRL therapy on peri-operative and long-term outcome is not clear. Although improvement in short-term remission rate was reported for macroadenomas and invasive adenomas, there was no significant change in the long term. No favourable effect on cardiac function, anaesthetic risks, surgical outcome or hospital outcome has yet been demonstrated, however selected cases might benefit clinically.84–87
Radiation therapy remains third in line in the treatment algorithm for acromegaly, and is usually considered in cases of large residual (or enlarging) tumour following surgery, and if medical therapy is unsuccessful or not tolerated.13 Stereotactic techniques, stereotactic radiosurgery (SRS) and fractionated stereotactic radiotherapy (FSRT) provide more precise, higher radiation dose to the targeted tumour, and limit irradiation to the adjacent normal structures compared to conventional radiotherapy (CRT). In a recent systematic review, the efficacy of SRS was similar to CRT, but has a potentially higher rate of biochemical control and lower rate of hypopituitarism.88 Favourable prognostic factors for efficacy of SRS include a higher margin radiation, higher maximum dose and lower initial GH/IGF-1 levels.89 The main side effect of SRS is radiation-induced hypopituitarism, while optic neuropathy, cranial neuropathies, brain radionecrosis and cerebrovascular disease occurred infrequently.90 FSRT appears to have a similar efficacy and risks with SRS except for the stroke risk that seems to be higher with FSRT.90,91
Conclusion
Novel insights into the diagnosis and pathophysiology of acromegaly have been acquired over recent years. Increased awareness of assay technical issues and discordant results affecting the biochemical assessment of the disease will ensure prompt diagnosis and initiation of treatment. Expanded knowledge on the histological and molecular levels, and development of new markers of response and resistance to SRL emphasised the importance of a personalised approach rather than following a universal algorithm for therapy. Future studies are needed to clarify the role of innovative formulation, combination and peri-operative pharmacotherapy on disease remission rate and patient QoL. The treatment of acromegaly remains multimodal for most patients and a multidisciplinary team is essential for optimal disease management and outcome.