In the adult male, hypogonadism is usually defined by the failure of the testis to produce physiological concentrations of testosterone and/or a normal number of spermatozoa.1 When due to abnormalities of the hypothalamic-pituitary axis, the condition is referred to as hypogonadotropic or central hypogonadism, whereas those due to testicular defects are called hypergonadotropic or primary hypogonadism. However, these definitions are inadequate for most paediatric conditions. Indeed, during the largest part of infancy and childhood, the hypothalamic-gonadotroph axis is quiescent and the testes produce neither spermatozoa nor detectable amounts of testosterone. A more adapted definition and classification of male hypogonadism is based on the developmental physiology of the gonadal axis.2 Accordingly, the definition should be extended to any decreased testicular function as compared to what is expected for age, involving impaired hormone secretion by Leydig cells (i.e., androgens, insulin-like 3 [INSL3]), Sertoli cells (i.e., anti-Müllerian hormone [AMH], inhibin B) and/or a disorder of spermatogenesis. Moreover, it should be emphasised that primary hypogonadism rarely presents with elevated gonadotropin levels3 and even anorchidism might present with normal gonadotropin levels in about 30–60% of the cases during childhood,4 indicating that the term hypergonadotropic hypogonadism may be misleading in paediatrics. Additionally, since circulating gonadotropin concentrations are low in the normal prepubertal boy, it is difficult to demonstrate the existence of levels below normal, thus making the term hypogonadotropic hypogonadism also inadequate.
A clear understanding of the developmental physiology of the hypothalamic-pituitary-testicular axis is essential for the comprehension of the pathogenesis of hypogonadal states in paediatric ages (Figure 1). Under hypothalamic control, the gonadotrophs secrete luteinizing hormone (LH) and follicle-stimulating hormone (FSH) during the second and third trimesters of foetal life, and also during the first 3–6 months after birth – a period usually referred to as mini-puberty.5,6 LH induces Leydig cell activation, resulting in testosterone production at levels that are similar to those of the adult. Androgens are responsible for testicular descent and penile growth. FSH induces Sertoli cell proliferation7 and since Sertoli cells represent the largest part of testicular mass before puberty, FSH is the main factor regulating testicular volume (Figure 2). Sertoli cells produce peptide hormones, such as inhibin B and AMH. Although their basal production is gonadotropin-independent, there is clinical8–10 and experimental evidence11,12 that FSH increases Sertoli cell secretion of inhibin B and AMH.
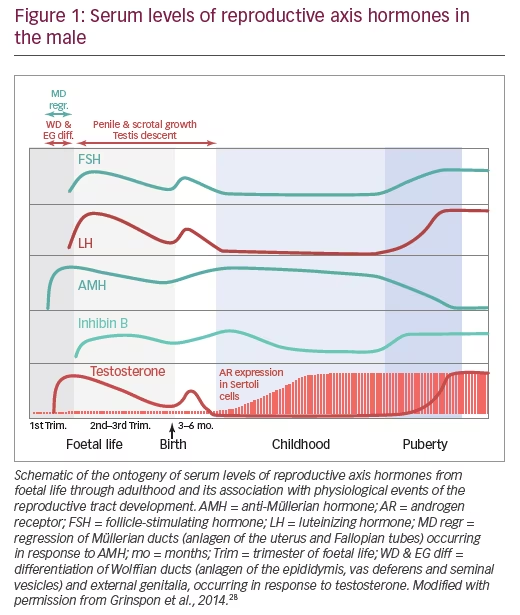
Around birth, there is a transient decrease in gonadotropin and testicular hormone levels,13 but from the second week of postnatal life, all hormone levels increase and remain high for 3–6 months.6 Thereafter and during the rest of infancy and childhood, LH and testosterone decrease to very low or undetectable levels. FSH and inhibin B also decrease but remain clearly detectable.14 Conversely, AMH peaks in the first year and remains high during childhood15,16 until it decreases in puberty inhibited by testosterone.17 In spite of the high levels of testosterone that seminiferous cords are exposed to during the foetal and postnatal periods, no signs of maturation are observed in Sertoli and germ cells. This is explained by the fact that Sertoli cells are physiologically insensitive to testosterone because the androgen receptor is not expressed in Sertoli cells until the second year of postnatal life (Figure 1).18 In fact, when elevated androgen levels abnormally persist beyond the age of 1 year (e.g. in boys with central precocious puberty), clear signs of Sertoli cell maturation appear.19
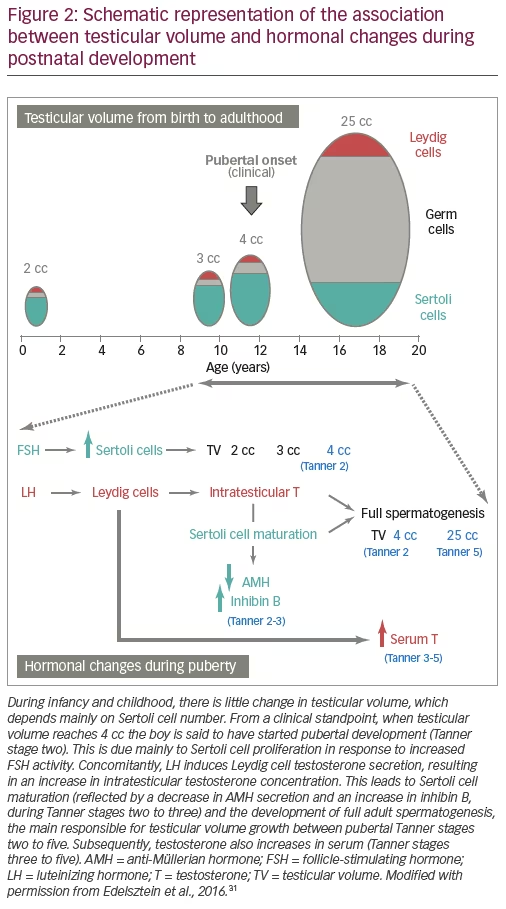
The onset of puberty is characterised by the increase in gonadotropin pulse frequency and amplitude. FSH further boosts testicular volume to >4 cc (Figure 2). LH reawakens Leydig cell activity, leading initially to an increase in intratesticular testosterone concentration which provokes Sertoli cell maturation: they stop dividing and decrease their expression of AMH.5 Mature Sertoli cells are able to sustain spermatogenesis;20 the completion of this process leads to sperm production and also results in the remarkable increase of testicular volume to 15–25 cc. The elevation of serum testosterone and the resulting development of secondary sexual characteristics is a relatively late event in boys.
Central hypogonadism in boys
Aetiology
Congenital central hypogonadism
Congenital central (“hypogonadotropic”) hypogonadism reflects the failure of the pituitary to produce sufficient amounts of gonadotropins. Most frequently, it is caused by deficient production, secretion or action of gonadotropin-releasing hormone (GnRH).21 The condition can be isolated and it is known as isolated hypogonadotropic hypogonadism,22 or associated to other pituitary hormone deficiencies.23 Several gene defects have been described as the cause of gonadotropin insufficiency. The pathogenesis may be related to: 1) altered GnRH neuron formation in the olfactory placode or its migration to the hypothalamus (usually associated with hyposmia/anosmia and known as Kallmann syndrome), 2) abnormal GnRH production due to defects in regulatory factors or in the GnRH1 gene, and 3) defective action in the gonadotroph owing to abnormalities in the GnRH receptor or its transduction pathway.21,22 Extremely rare are mutations in the genes encoding the beta subunits of LH or FSH.24 Finally, genes associated with defective formation of the hypothalamic-pituitary region have been described in a low proportion of cases with multiple pituitary hormone deficiency.25
Acquired central hypogonadism
Of all paediatric and pubertal forms of central hypogonadism, approximately 20% are of functional origin. In these cases, a chronic illness or an underlying stressor causes the suppression of gonadotropin secretion and the condition can be reverted when the chronic illness is adequately treated.26 Conversely, permanent forms are due to conditions affecting the hypothalamic-pituitary area, e.g. high radiation doses, tumours, infections, surgery, etc.
Clinical presentation
Neonatal and infancy
The insufficiency of LH and testosterone during the second half of foetal life usually results in micropenis, cryptorchidism and hypoplastic scrotum. On the other hand, FSH insufficiency leads to microorchidism due to Sertoli cell hypoplasia. Midline defects and signs like hypoglycaemia or jaundice can orientate to multiple pituitary hormone deficiencies. Unfortunately, the insufficient attention given to cardinal features, such as micropenis, cryptorchidism, cleft lip/palate, deafness, coloboma, etc., in the newborn or infant results in missing the window of opportunity for the diagnosis during the postnatal activation period.
Prepubertal age
Since changes in testicular volume and penile size are minimal during childhood, acquired central hypogonadism established in this period of life may go underdiagnosed even if the underlying aetiology is evident, e.g. tumours or surgery of the central nervous system.
Pubertal age
Absence of pubertal signs is the main feature of complete forms of central hypogonadism, whereas delayed onset or progression, or even arrest of pubertal development, may be indicative of partial forms. At this age, smell testing can be helpful if a congenital form (Kallmann syndrome) is suspected.
Hormonal laboratory testing
Gonadotropins and testosterone
The window of opportunity for confirming central hypogonadism using gonadotropin and testosterone measurements in term newborns and infants resides between the third week and the third month. Indeed, in the first 2 weeks, gonadotropins and testosterone may still be low and only starting to increase after the perinatal nadir in normal newborns,13 thus hormone values should be interpreted cautiously in order to avoid false positive diagnoses. Similarly, from the fourth to sixth month onwards, gonadotropins and testosterone may already be decreasing physiologically. During the post-natal active period, or mini-puberty, low LH and testosterone have high positive predictive values for central hypogonadism.23,27–29
If the diagnosis was not certified before the sixth month of age, LH and testosterone levels are no longer informative until pubertal age. Although FSH is usually neglected in the study of male central hypogonadism, circulating levels never drop to undetectable during childhood and can therefore be more informative than LH. Indeed, in a series of boys presenting without spontaneous pubertal development, basal FSH had a higher predictive value than LH for the diagnosis of central hypogonadism.30
Sertoli cell markers
As already mentioned, AMH and inhibin B are Sertoli cell products, whose basal production is independent of gonadotropins. Nevertheless, FSH is capable of increasing inhibin B8,9 and AMH9,10 testicular secretion, thus indicating that they both are good markers of FSH action on the testis.31 The increased AMH testicular output in response to FSH in prepubertal male patients reflects two effects of FSH on immature Sertoli cells. On one hand, FSH induces Sertoli cell proliferation,7 which explains that FSH treatment in newborns or infants causes an increase in testicular volume.9 AMH levels reflect the amount of functional Sertoli cells in different conditions.32–34 On the other hand, using two experimental models, knockout mice null for the gene encoding the FSH beta subunit12 and the prepubertal Sertoli cell lineage SMAT1 expressing AMH,35 we showed that FSH upregulates the AMH promoter activity signalling through the cyclic AMP-mediated pathway, mainly via protein kinase A activity.36,37
The aforementioned regulation of Sertoli cells by FSH during foetal life and the neonatal period explains the findings of microorchidism and low serum AMH and inhibin B in boys with congenital central hypogonadism.8,9,23,29,38
Treatment
Testosterone
In patients with congenital central hypogonadism, the presence of micropenis may prompt testosterone treatment. When the diagnosis is made at birth – or more rarely during childhood – depot testosterone (testosterone enanthate or cypionate) at 25–50 mg/dose intramuscular (IM) every 4 weeks for 3 consecutive months may be sufficient to normalise penile size without provoking an advancement of bone age.39 In older boys with central hypogonadism reaching the age of puberty, steroid replacement is necessary for the development of secondary sex characteristics, the acquisition of full bone mass and the optimisation of height velocity, as well as for avoiding psychosocial distress. Depot testosterone is given at 50 mg IM each month for 6–12 months, with a subsequently progressive dose escalation until a full dose of 250 mg/month in 2–3 years. The aim is to mimic normal pubertal tempo. When full pubertal development is attained, treatment should be switched to follow standards for adults.1 Alternative approaches include oral androgens such as testosterone undecanoate40 and transdermal gels.41,42
Gonadotropins
Exogenous testosterone supplementation aims to attain physiological circulating androgen levels. However, physiological intratesticular testosterone concentration is never reached with these treatments, whose disadvantages are that Sertoli cells neither proliferate nor mature and, subsequently, adult spermatogenesis does not develop. In fact, testicular volume remains small (<4 cc) in patients receiving only testosterone treatment.
Treatment with pulsatile GnRH or combined gonadotropins is well established in adult patients with central hypogonadism seeking fertility.21 Conversely, limited information is available on the use of gonadotropins in paediatric patients.43 Observational studies on infants with congenital central hypogonadism suggest that the postnatal peak in reproductive hormones plays an important role in genital development.44 Therefore, various authors have proposed the use of gonadotropins during infancy, instead of testosterone, to mimic mini-puberty in patients with congenital central hypogonadism. A case report describes an infant treated from the age of 7.9 months with recombinant human (rh) LH (subcutaneous injections 20 IU twice a week) and FSH (subcutaneous injections 2.5 IU/kg twice a week).45 Testicular volume increased 170%, reflecting FSH action on Sertoli cells.
A case series reports eight patients aged 0.25–11 months who received continuous infusion of rhLH and rhFSH with insulin pumps at a daily rate of 50 and 75–150 IU, respectively.9 Testicular hormones reached “mini-puberty” levels, complete testis descent occurred in six patients and partial descent in two, and testes and penis reached normal dimensions in all cases. Similar results were observed in patients receiving continuous subcutaneous infusion of rh gonadotropins.46 Even though long-term follow-up is needed to fully evaluate the outcomes of this therapeutic approach, these results suggest that subcutaneous gonadotropin infusion is able to induce not only Sertoli cell proliferation and function but also testis descent in patients with congenital central hypogonadism, thus avoiding surgical treatment.47
Only one study reports prepubertal treatment with rhFSH of three boys with central hypogonadism.8 In this case, the rationale was that FSH would be capable of inducing Sertoli cell multiplication before they mature at puberty. The boys were given rhFSH subcutaneously (1.5 IU/kg) three times a week for 12 months. Treatment induced testicular growth and inhibin B production, reflecting stimulation of immature Sertoli-cell function. These findings suggest that FSH treatment in childhood is capable of inducing Sertoli-cell proliferation, which may result in an increased sperm-producing capacity in adulthood.
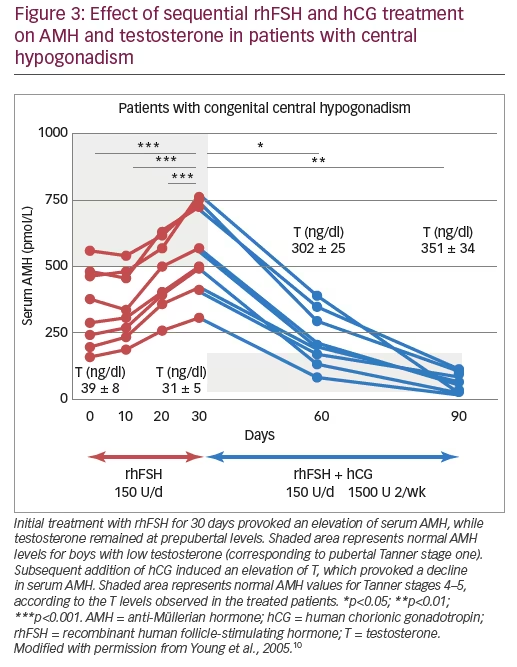
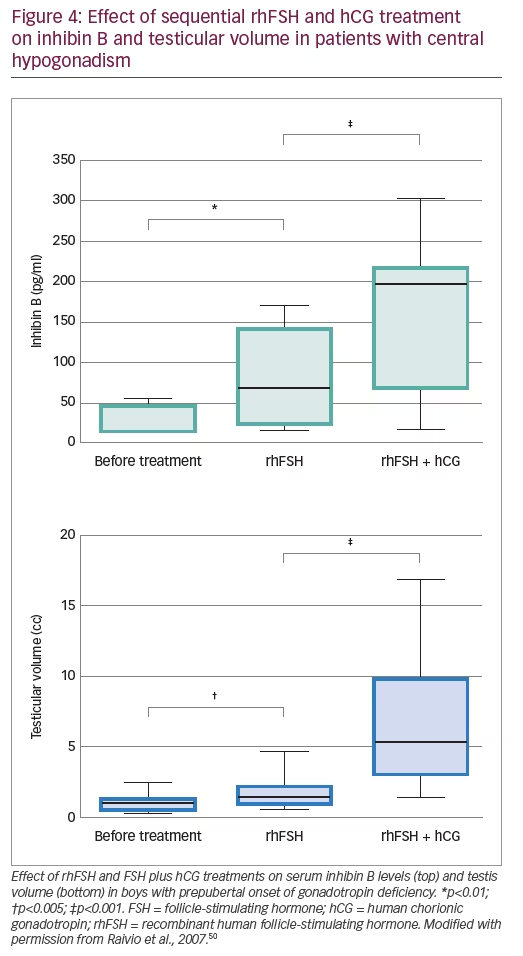
Finally, several publications exist on combined recombinant gonadotropin treatment for the induction of puberty in boys with central hypogonadism. Some of them used the standard protocols for adults initiating with human chorionic gonadotropin (hCG) and then adding rhFSH48,49 and showed acceptable results. Moreover, in studies using initial priming with rhFSH prior to induction of puberty with the combination of rhFSH and hCG, rhFSH induced prepubertal testicular growth and increased serum levels of AMH (Figure 3)10 and inhibin B levels (Figure 4),10,50 confirming the importance of FSH action on immature Sertoli cells. The addition of hCG resulted in a progressive increase in testosterone production, Sertoli cell maturation suggested by an important decrease in serum AMH and acquisition of sperm production.10 Also, the increase of inhibin B proved useful for monitoring spermatogenic development.50 More recently, an open-label randomised study in hypogonadal males aged >18 years showed better results in men receiving rhFSH pre-treatment in terms of spermatogenic development.51
Concluding remarks
While LH and testosterone levels have been used for years in the diagnosis of central hypogonadism at pubertal age and adulthood, in paediatric patients they are useful only during the early post-natal activation period. During childhood, low levels of the Sertoli cell markers AMH and inhibin B support the diagnosis of central hypogonadism. Furthermore, although testosterone replacement therapies have been used for many years with satisfactory results on the development of secondary sex characteristics in adolescents with central hypogonadism, more recently the attention has been driven to the physiological importance of the intratesticular actions of FSH and testosterone during early infancy and the first stages of pubertal development.
The limited number of studies available to date demonstrate that a sequential treatment approach with rhFSH priming to induce Sertoli cell proliferation, before adding hCG or rhLH to provoke an increase in intratesticular testosterone concentration and Sertoli cell maturation, shows encouraging results in terms of induction of testicular growth and sperm production.8,10,50,51 To definitively prove the superiority of this approach, larger randomised prospective studies are necessary.