The overall association between obesity and type 2 diabetes is so linear and strong that a causal relationship between the two has traditionally been assumed.1 There are some questions, however, yet to be answered.2 The association of diabetic complications with obesity has consistently shown paradoxical results.3,4 In people with diabetes with a higher body mass index (BMI) up to class 1 (BMI <35 kg/m2), obesity seems to have a protective effect.5 There is no satisfactory explanation for this reverse epidemiology phenomenon. The aim of this review is to attempt to generate a unifying hypothesis explaining this and other paradoxical phenomena and at the same time critically appraising the complex relationship between diabetes and obesity.
Epidemiological data
Obesity and diabetes
Increased body weight has been positively correlated with increased risk of diabetes. In data from NHANES 3, increasing BMI was shown to increase the risk of diabetes in a linear fashion.6 Similarly, in the UK, 90% of people suffering from type 2 diabetes mellitus are either obese or overweight.1 In this study, obesity conferred a fivefold higher risk of developing diabetes compared with those with a normal weight.1 It was, however, also shown that only 12.4% of all obese people would eventually develop diabetes. Hence, it seems that, obesity, on its own, is not enough to cause type 2 diabetes mellitus.
Data from Southeast Asia also shows that people with obesity are more likely to develop diabetes compared with a healthy body weight, even though Southeast Asians have a lower prevalence of obesity (by BMI criteria), both in the general population and among people with diabetes.7,8 However, this population is shown to have higher insulin resistance and impaired glucose tolerance even at lower BMIs.8,9,10 This paradox has been explained by demonstration of more visceral fat in this group.11,12 This concept has already been challenged by other studies, which demonstrate that upper body subcutaneous fat is the predominant contributor towards the systemic free fatty acid pool and that visceral adipose tissue plays a relatively minor role in the entire process.13–16 However, despite the contradictory results from these studies, literature from all regions of the world shows significant associations between prevalence of obesity and diabetes. Hence, a causal relationship has been naturally assumed.
Obesity and diabetes complications
Contrary to the association of obesity with diabetes onset, its association with diabetic complication seems to be paradoxically inverse and, hence, protective.5,17,18 In a large meta-analysis from Scotland, 29,316 overweight/obese people with type 2 diabetes mellitus were analysed in terms of cardiovascular complications.17 It was concluded that within patients with BMI up to 40 kg/m2, there was no difference in the incidence of all-cause mortality, myocardial infarction, congestive heart failure, peripheral vascular disease or cerebrovascular disease.17 Only when BMI was more than 40 kg/m2, an increase in the incidence of all-cause mortality, congestive heart failure and cerebrovascular disease was found.17 In another study published in the Journal of American Medical Association, data from five prospective heart cohorts was analysed to establish a relationship between BMI at diabetes onset and all-cause mortality, cardiovascular mortality and non-cardiovascular mortality on a subsequent follow up.18 In all cohorts combined, 89% of people who developed diabetes were overweight or obese.18 However, interestingly, when data of these overweight/obese patients with diabetes were compared with that of healthy weight patients with diabetes, it was shown that overweight/obese patients with diabetes had a significantly lower all-cause mortality and non-cardiovascular mortality while cardiovascular mortality was non-significantly (p=0.06) lower.18
In the largest meta-analysis to date on this subject, 16 cohort studies on all-cause mortality (n=445,125) and two studies on cardiovascular mortality (n=92,841) among people with diabetes were evaluated.5 A non-linear association was observed between BMI and all-cause mortality among patients with type 2 diabetes, with the lowest overall mortality reported at a BMI range of 28–30 kg/m2. Every 5 kg/m2 increase in BMI was significantly associated with further decreased all-cause mortality (HR = 0.99, 95% confidence interval [CI]=0.97–1.00, p=0.04).5 Lowest cardiovascular mortality was reported at BMI range of 29–31 kg/m2 amongst people with diabetes.5 In light of the above literature, it is difficult to deny this obesity paradox in diabetes, which seems to confer a survival advantage.
Current pathophysiological considerations
The current belief in the scientific community is that obesity induces insulin resistance and, hence, is causally related to the onset of diabetes. But, subsequent development of chronicity with diabetes, seems to lead to the same adiposity becoming protective (obesity paradox).19 However, to date, there has been no satisfactory explanation of why a higher BMI protects against all-cause mortality in diabetes, and we are still yet to discover when and how the relationship between obesity and diabetes changes from one that is seemingly causative to one that appears to be protective. Another factor that needs to be considered is that not all obese patients eventually go on to develop diabetes.1 Challenging this concept is important for the future of diabetes medicine as we are already witnessing the promotion of weight loss medicines/procedures as anti-diabetes therapies. Moreover, as worldwide obesity rates soar, figuring out the exact relationship between weight and diabetes is extremely important to individualise risk calculation as well as preventive and therapeutic interventions.
Uniform hypotheses
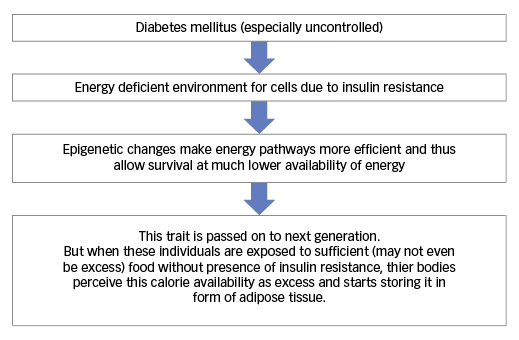
Body weight is regulated by the human body through a lot of short- and long-term mechanisms that include a variety of hormones such as leptin, ghrelin, insulin and cholecystokinin as well as neural hypothalamic circuits.20 The increasing worldwide prevalence of obesity has been attributed to availability of excess food as well as lack of physical activity. However, it is a common clinical observation that, despite the same overall hormonal and neural controls and the same calorie composition of diet and level of exercise, different individuals behave differently in relation to weight balance, with some people losing weight while others gain weight. It is also noted that people who keep putting weight on have a strong background of diabetes in their families.21 We propose that the progeny of people with diabetes develop more energy efficient metabolic pathways, which might allow their cells to survive even in situation of lower glucose availability. These changes may be mediated via undefined epigenetic changes and may be passed on from one generation to the next.22,23 These changes would enable the progeny of people with diabetes to have a much lower basal requirement of calories for survival. However, when these individuals are exposed to environments with a sufficient availability of food (even without the presence of insulin resistance), this survival advantage may become maladaptive and they may start gaining weight at a much lower calorie intake. This may explain the increasing prevalence of obesity within the progeny of people with diabetes (see Figure 1) and we propose to call it ‘transgeneration energy karma’ theory.
Figure 1: Transgeneration energy karma – possible explanation for increased risk of obesity within progeny of people with diabetes
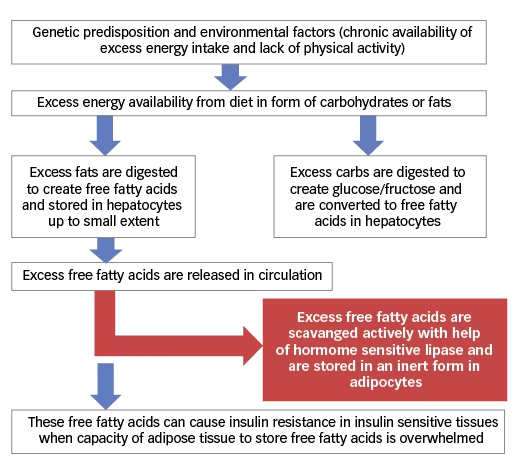
The human body utilises glucose as its primary source of energy. However, glucose is not suitable as energy storage on long-term basis or transport of energy between organs. Free fatty acids are energy-dense molecules and seem to be ideal for this purpose as they are an efficient way to transfer energy between different organs. In the presence of excessive energy molecules, the liver does store a limited amount of excess calories in the form of glycogen. The rest of the excess energy, either in the form of glucose or free fatty acids, is converted into triglycerides. A small fraction of these triglycerides is stored intra-hepatically in hepatocytes, whereas a larger majority are transported out in the form of chylomicrons. From the circulation, these are scavenged into adipose tissue by hormone sensitive lipase (see Figure 2). Adipose tissue is the only tissue where free fatty acids can be stored safely for a long time.23,24 Free fatty acids, within adipocytes in peripheral insulin-sensitive tissues, induce insulin resistance by various mechanisms.
Figure 2: Relationship of excess energy intake, free fatty acid metabolism and insulin resistance
Initially, it was thought that free fatty acids impair glucose utilisation within tissues by the accumulation of acetyl coenzyme A (acetyl-CoA), which in turn inhibits pyruvate dehydrogenase and phosphofructokinase activity.25 However, it has been shown that the primary defect actually lies in glucose uptake and not in glucose oxidation.26 Molecular mechanisms involved in this phenomenon of impaired glucose transporter type 4 (GLUT-4) translocation to plasma membrane of insulin-sensitive cells have been well delineated. These mechanisms have been discussed in detail elsewhere.16,24,26,27
In response to chronic overfeeding, free fatty acids are actively extracted from the circulation and deposited within adipocytes in the form of triglycerides, under the influence of insulin. However, these free fatty acids do not induce insulin resistance in adipose tissue initially as they are rapidly converted to neutral triglycerides. As more and more triglycerides get stored, adipocytes undergo hypertrophy followed by hyperplasia. Hence, while adipocytes play an important role by extracting free fatty acids from circulation and preventing insulin-sensitive tissues from becoming exposed to them, when the capacity to store triglycerides is saturated in adipocytes, free fatty acids within the circulation can, and will, induce insulin resistance within insulin-sensitive tissues.27,28 Hence, we hypothesise that the gain of adipose tissue is an active protective mechanism to delay the development of insulin resistance in response to chronic overfeeding.
If we accept this hypothesis, the question that arises next is that if obesity is a normal protective response of the human body against insulin resistance then why does it not happen in all people with diabetes and in different ethnicities similarly? An explanation is that, as adipocytes are exposed to chronic excess of free fatty acids, the capacity to store free fatty acids may vary in different individuals and different racial backgrounds. Differences in the capacity of individual adipocytes to store a certain amount of fat (capacity for hypertrophy), as well as the capacity to undergo cell division (hyperplasia), may be responsible for this variation. This may explain the occurrence of diabetes in Asians at a much lower BMI as well as at an earlier age. This has been termed as the ‘adipocyte overflow hypothesis’.29,30 Sniderman et al. proposed that as obesity develops, South Asians exhaust the storage capacity of their superficial subcutaneous adipose tissue compartment before Caucasian populations do and that leads to an early expansion of visceral adipose tissue, which, in turn, leads to the earlier development of metabolic complications.29 Adipocytes isolated from Asians have been shown to be larger in size and smaller in number compared with Caucasians.29,31
Certain adipokines have also been shown to affect insulin resistance directly. Larger adipocytes have also been demonstrated to have higher pro-inflammatory cytokines, relative ischaemia, lower secretion of adiponectin and higher levels of resistin.32 However, which one of these is a primary phenomenon is still being debated. Meanwhile, as insulin resistance increases within adipose tissue, the lipogenic potential of adipocytes also becomes limited and lipolysis is favoured over lipogenesis more and more. Hence, free fatty acids are released into circulation, thus, establishing a vicious cycle.28
In a recent study from Scotland and England, diet intervention aimed at making people lose weight and evaluating its impact on ‘reversal’ of diabetes has been shown to very effective.28,33 Diabetes remission in this study was defined as glycated haemoglobin (HbA1c) <6.5% for at least 2 months without any anti-diabetic medication. Individuals (n=306), aged between 20–65 years, with diabetes of <6 years’ duration and with a BMI 27–45 kg/m2 were recruited. Participants were provided with approximately 850 Kcal/day in form of meal replacements to start with and normal diet was introduced subsequently in a stepped manner. At end of 1 year of intervention, 46% of people achieving remission was strongly correlated with weight loss. A loss of 15 kg or more resulted in remission in 86% of people.33 This study is a significant step towards explaining that excess energy availability is causative factor for the occurrence of diabetes and restricting calories may actually reverse it. Also, the fact that people needed to be on much lower calories supports the ‘transgenerational energy karma’ theory.
Classification of obesity into metabolically healthy obesity and metabolically unhealthy obesity can also be explained by this hypothesis. A person with a higher BMI would still be metabolically healthy until the time adipocytes are able to store excess free fatty acids. When capacity of adipocytes to store free fatty acids is overwhelmed, metabolic abnormalities ensue. As the capacity to store fat is different for each individual, it does not confine to the boundaries of BMI.
Adipose tissue not only extracts harmful fatty acid from circulation but is also associated with the secretion of soluble tumour necrosis factor alpha (TNFa) receptors into the circulation that then bind excessive TNFa present in circulation, produced as a result of any chronic disease including diabetes.4 An increased lipoprotein concentration in obesity has also been proposed to bind and clear endotoxins from circulation.34 These explanations, among others, have been suggested to be responsible for the increased survival within people with chronic diabetes as a result of diabetes itself or independently in association with other chronic diseases, such as coronary artery disease, stroke, chronic kidney disease, chronic obstructive lung disease and infections.4,34–36
Beta cell dysfunction has also been proposed as a prerequisite for the development of diabetes as not all people with obesity eventually develop diabetes.37 Beta cell mass is higher in obese people with diabetes compared with people with normal weight diabetes.8 As both obese and lean people with diabetes have insulin resistance, different beta cell mass in these two groups cannot be attributed to insulin resistance directly. Other signalling molecules may mediate this beta cell expansion. G-protein coupled receptor signalling, as well as increased secretion of hepatocyte growth factor, has been proposed as potential links among others.38
Conclusion
Adipose tissue expansion is an active phenomenon at the onset of diabetes but seems to confer protection from development of diabetic complications as the disease starts to become chronic. Reduced energy requirement for cell survival may be the starting point in a self-perpetuating cycle of increasing prevalence of obesity and diabetes. In the presence of chronic excess of food, the human body tries to store harmful free fatty acids in a neutral form (triglycerides) within adipose tissue.
A differential capacity to store amounts of free fatty acids may be responsible for ethnic and individual differences in the relationship between adiposity and diabetes. Increase in the amount of fat stored in adipocytes alters their metabolism and energy homeostasis. Fewer free fatty acids are cleared from circulation and more free fatty acids are released into circulation from adipose tissue as its capacity to store triglycerides is saturated. Pro-inflammatory cytokines, relative hypoxia, lower secretion of adiponectin and increasing insulin resistance within adipose tissue may be responsible for a lipolytic shift in adipose tissue. Moreover, obesity has been strongly associated with the expansion of beta cell mass in rodent as well as human studies.39–43 Other mechanisms proposed to explain increased survival in people with higher BMI and chronic complications of diabetes include increased secretion of soluble TNFa receptors and lipoproteins from adipose tissue. Hence, this hypothesis offers an overarching and effective explanation for the noted paradoxes of diabetes epidemiology and obesity. ■