




Oncocytomas are epithelial tumours composed of cells with eosinophilic, mitochondria-rich cytoplasm.1 They can occur in various organs, such as the kidneys, thyroid, pituitary gland, salivary glands and parathyroid glands. Rarely, it has been reported in the respiratory and gastrointestinal tracts.2–4 Adrenal oncocytomas are extremely rare, with nearly 200 published cases since Kakimoto et al. reported the first case in 1986.5,6
The detection of adrenal oncocytomas is mostly incidental since they are frequently non-functional; however, biochemical analyses are recommended to exclude functional status.7 Imaging is not a reliable method of diagnosis for oncocytomas as they can display features often present in other tumours, such as carcinoma; histological analysis is required for an accurate diagnosis.7 Since oncocytomas are benign lesions associated to favourable prognosis, the diagnosis has implications for follow up. Despite the impermissible indication for surgery, there are scarce data concerning follow-up strategies.7,8
Case presentation
We present a 55-year-old male patient with dyslipidaemia and important ethylic habits. He is a former smoker with a medical history of sacral-coccygeal cyst, traumatic amputation of two left-hand fingers in 2000, a peroneal fracture in 2014 and is medicated with simvastatin 20 mg daily. In 2017, the patient complained to his general physician of abdominal pain. Abdominal ultrasound revealed a right adrenal lesion, further characterized with a computed tomography (CT) scan that showed a nodular heterogeneous lesion measuring 49 x 64 x 56 mm with a calcification focus in the right adrenal gland. In this context, the patient was referred to the general surgery and endocrinology department in July 2017.
During the endocrinology appointment for an adrenal incidentaloma, we requested a hormonal study to evaluate the possibility of a functional mass and a renal CT scan. Since the patient did not have hypertension, we focused on pheochromocytoma and Cushing’s syndrome exclusion. The results of this study (in May 2018) were: 1 mg overnight dexamethasone suppression test of 2.9 µg/dL (normal range <1.8 µg/dL), adrenocorticotropic hormone (ACTH) of 22 pg/mL (<46 pg/mL), dehydroepiandrosterone sulfate (DHEA-SO4) of 699 µg/dL (80–560 µg/dL), testosterone of 167 ng/dL (241–827 ng/dL), estradiol of 83.1 pmol/L (<146.1 pmol/L) and normal urinary metanephrines. A renal CT scan confirmed a right adrenal mass of 49 x 64 x 56 mm (Figure 1).
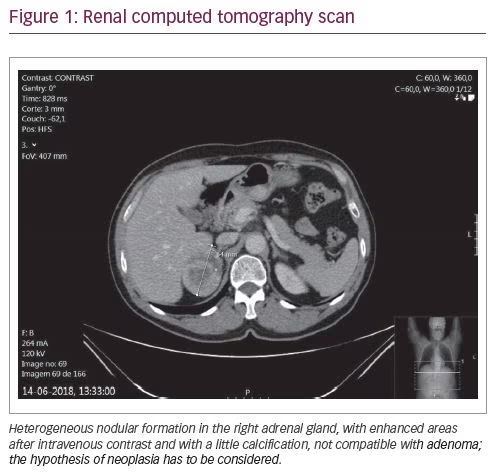
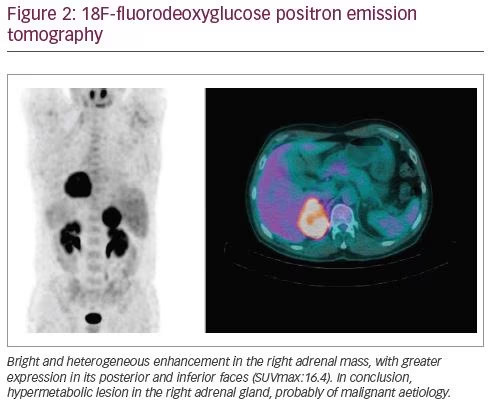
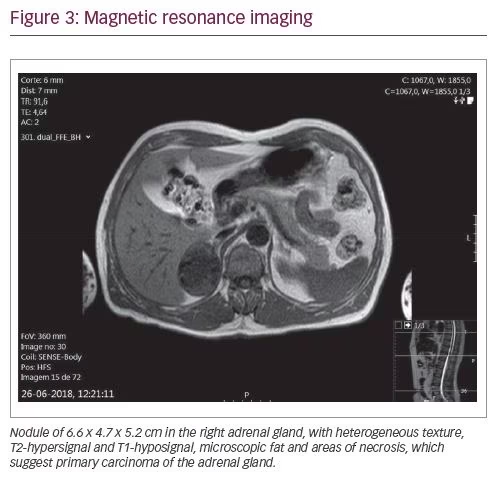
The patient was evaluated by the general surgery department and he underwent 18F-fluorodeoxyglucose positron emission tomography (FDG-PET) and magnetic resonance imaging (MRI) for better assessment of the nature and limits of the lesion. The FDG-PET (Figure 2) showed an 18F-FDG hypermetabolism in the right adrenal mass (SUVmax: 16.4) and the MRI (Figure 3) detected a nodule of 66 x 47 x 52 mm in the right adrenal gland, without invasion signs.
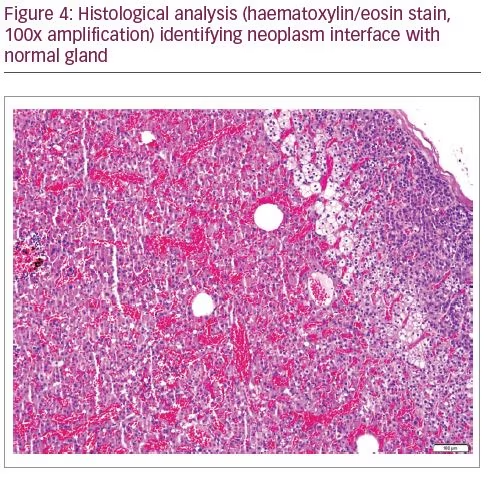
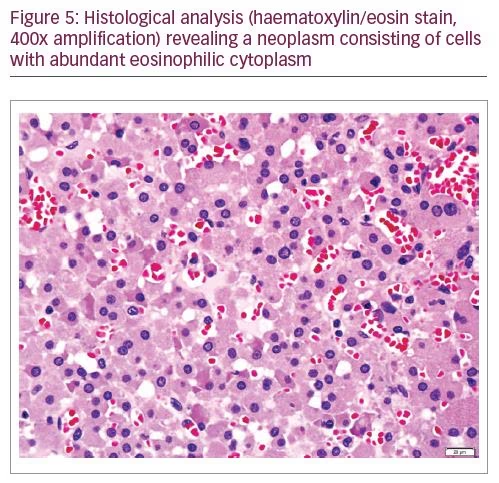
In August 2018 the patient underwent right adrenalectomy by laparotomy with the collaboration of the adrenal and hepatobiliary surgical teams. To prevent postoperative adrenal insufficiency, the patient was medicated with hydrocortisone. He recovered uneventfully. The operative piece weighed 124 g, measured 9.5 x 5.5 x 5.0 cm, with a nodule of 6.0 x 5.5 x 5.0 cm of well-defined limits, spongy, brownish, with haemorrhage areas. The histological analysis (Figures 4 and 5) revealed the diagnosis of adrenocortical neoplasm with adenoma features with predominance of oncocytic cells (adrenocortical oncocytoma).
One month after surgery, the patient was asymptomatic, without signs of a surgical scar infection or incisional hernia. Four months after adrenalectomy, the general surgery team scheduled the next evaluation at 1-year post-operation, due to the patient’s positive recovery. In February 2019, the patient was evaluated in the endocrinology department and displayed normalised analytical parameters: cortisol 13.1 µg/dL (4.3–22.4 µg/dL) and DHEA-SO4 120 µg/dL (80–560 µg/dL). He is currently in follow up.
Discussion
Oncocytic cells were first described in 1931 by H Hamperl as cells with eosinophilic and granular cytoplasm.9 Recent advances have allowed us to understand that these features were a result of numerous mitochondria.9 Oncocytic tumours arise frequently from the kidneys, representing 3–7% of all renal neoplasms,7 but are already found in thyroid, pituitary gland, salivary glands, parathyroid glands, respiratory and gastrointestinal tracts.2–4 Adrenal oncocytomas are extremely rare, with nearly 200 published cases since Kakimoto et al. reported the first case in 1986.5,6 The mechanisms for oncocytosis are not well understood and there are two theories: the first defends that mitochondria proliferation is a result of a genetic mutation and the second that it is a consequence of an epigenetic event resulting from cellular hypoxia.10,11
Adrenal oncocytoma can occur in a large age range (15–77 years), more frequently in women (2.5:1.0), in the left adrenal gland (3.5:1.0), without known risk factors.7,8,12 Frequently, an adrenal oncocytic neoplasm is discovered incidentally and is a rare entity to consider in the differential diagnosis of an adrenal incidentaloma. Differential diagnoses include cortical adenoma, pheochromocytoma, Cushing´s syndrome, adrenocortical carcinoma, granulomas, adrenal cyst, myelolipoma, ganglioneuroma and metastatic disease.7 Although it is, by definition, non-functioning, recent data indicate that 17.0–31.5% of adrenal oncocytomas have hormone production.12 In our case report, 1 mg overnight dexamethasone suppression test revealed probable autonomous cortisol secretion. Adrenal oncocytoma has been diagnosed in the context of Cushing’s Syndrome, pheochromocytoma, aldosteronoma, pseudoprecocious puberty and virilising symptoms in females.7,8,12 The exclusion of pheochromocytoma is particularly important in the diagnosis of adrenal oncocytoma as it affects pre- and post-operative procedures.7
Imaging methods (e.g. CT, MRI) lack diagnostic capacity for adrenal oncocytoma as it is difficult to differentiate it from a malignant lesion.7,8 Imaging features of adrenal oncocytoma include a size of generally >6 cm, lipid-poor, a higher attenuation on CT ranging from 20–40 HU, and malignant process features such as fibrous encapsulation or heterogeneous contrast enhancement.7 According to the latest guidelines, an adrenal incidentaloma of >4 cm and with more than 10 HU is likely malignant and surgery must be considered.13 Despite this fact, an adrenal oncocytoma is typically benign.7 Recent studies report a 22% incidence of malignant oncocytomas.7,12 The diagnosis depends on the pathological and histological examination of the operative piece. Macroscopically, adrenal oncocytoma appears round, encapsulated, with an average diameter of 8 cm, dark brown in colour with haemorrhage and necrosis areas. Histologically it is mainly composed of oncocytic cells.7,8
The treatment of choice for any suspicious adrenal mass is adrenalectomy, which may be by laparotomy when handling big lesions, or by laparoscopy for smaller lesions and if imaging displays a well-encapsulated tumour without invasion.7
Prognosis is determined by the characteristics of the oncocytoma. Adrenocortical tumours can be defined using the Weiss criteria of malignancy,14 which was adapted by Bisceglia et al. in order to distinguish malignant oncocytic tumours.15 The Lin–Weiss–Bisceglia risk system includes: major criteria (mitotic rate of >5 mitoses/50 high power fields, atypical mitosis and venous invasion), minor criteria (>10 cm of diameter and/or >200 g of weight, necrosis, capsular or sinusoidal invasion) and definitional criteria (dominance of oncocytic cells, high nuclear grade and diffuse architectural pattern). The presence of one major criterion classifies the oncocytoma as carcinoma; the presence of one or more minor criteria classifies it as having uncertain malignant potential; and the absence of criteria indicates benign behaviour.8,15
Follow-up recommendations are scarce; on one hand there is no record of recurrence of benign or indeterminate potential oncocytomas after surgery; however, malignant oncocytomas have a 5-year survival rate of 50–60% after surgery. 7,8 Of nine malignant cases, five were declared disease-free at 6 months, while four exhibited recurrence. It is therefore advisable, in the absence of evidence, to maintain a regular follow up for a minimum of 5 years.7,8
Conclusion
The case of a 55-year-old male who presented with abdominal pain has been described here. Abdominal ultrasound revealed an adrenal heterogeneous lesion that measured 49 x 64 x 56 mm on CT scan. The patient was referred for right adrenalectomy by laparotomy under the suspicion of a carcinoma. Histological analysis revealed an adrenocortical oncocytoma. One-month post-surgery, the patient had recovered well and was asymptomatic.
A large (>4 cm), heterogeneous, lipid-poor (>10 HU) adrenal incidentaloma with heterogeneous contrast enhancement and necrosis areas is suggestive of adrenocortical carcinoma and surgery is mandatory. Plasma or urinary metanephrines are of particular importance as a pheochromocytoma diagnosis can affect pre-operative and post-operative management; however, a 1 mg overnight dexamethasone suppression test, sex hormone levels, steroid precursors and aldosterone/renin ratio in the presence of hypertension or unexplained hypokalaemia are recommended.
Since imagiological characteristics of an oncocytoma are indissociable from the ones of a carcinoma, the diagnosis is determined through histological analysis. It is by definition a benign lesion, although it can be classified as having uncertain malignant potential or as carcinoma, with important prognosis and follow-up implications.
Adrenal oncocytoma is extremely rare, with less than 200 published cases, which makes it difficult to study, determine prognosis and define follow-up strategies. This case is a contribution to the knowledge on adrenal oncocytomas, since there is scarce information based on singular experiences.


