




Acromegaly Epidemiology and Clinical Manifestations
Acromegaly Epidemiology and Clinical Manifestations
Acromegaly is an uncommon disorder characterised by the hypersecretion of growth hormone (GH) resulting in an increase in serum insulin-like growth factor-1 (IGF-1) levels. This condition is most often caused by a pituitary adenoma. The prevalence is estimated around 40–125 per million and the incidence is around three to four new cases per million.1 Two studies have suggested that the prevalence of acromegaly may be more widespread than previously estimated. In a study including 71,972 subjects, the prevalence of pituitary adenoma was more than 3.5–5 times than previously reported, whereas in an another study the prevalence of biochemical acromegaly by screening for elevated IGF-1 levels was even higher with 1,043 cases per million of subjects.2,3 The mean age at diagnosis is around 40 years. The clinical manifestations of acromegaly are varied and comprise enlargement of the extremities, soft tissue swelling, coarsening of facial features, prognathism, macroglossia, increase in ring and/or shoe size, arthritis, hyperhidrosis and diabetes.
Acromegaly Diagnosis Criteria
The current international consensus for the diagnosis of acromegaly is based on the inability to suppress serum GH to less than 1 μg/l after glucose administration (75 g is recommended), in conjunction with a clinical suspicion and high IGF-1 levels.4
The diagnosis is often preceded by around 10 years of active unrecognised disease.5 The clinical manifestations of acromegaly depend on the level of GH and IGF-1, tumour size, the age of the patient and the delay in diagnosis.
Treatment Options in Acromegaly
The aims of therapy in acromegaly are alleviation of symptoms, complete tumour removal and a decrease in morbidity and mortality.6 Complications linked to acromegaly are principally premature atherosclerosis, hypertrophic cardiomyopathy, diabetes, arthritis, sleep apnoea syndrome and polyps of the colon.7 The cause of death in acromegaly patients is cardiovascular disease in 60 % of patients, 25 % respiratory disease and 15 % neoplasia.8
A biochemical complete control is achieved by a serum GH level <1 μg/l with a sensitive immunoassay or <2.5 μg/l with a sensitive enzyme-based immunoassay, normalisation of serum IGF-1 levels compared with age- and sex-matched controls and/or GH level under 0.4 μg/l after an oral glucose tolerance test (OGTT).4
Management of a patient with acromegaly requires a multidisciplinary collaboration with a general practitioner, endocrinologist, neurosurgeon and radiotherapist.
Primary surgical removal of the tumour is considered as the preferred treatment for acromegaly according to the guidelines of the American Association of Clinical Endocrinologists.4 Transsphenoidal surgery in particular is considered as the first approach in patients with microadenomas and in patients with macroadenomas that are associated with a mass effect. Surgical excision achieves a normalisation of serum IGF-1 in 56–68 % patients with non-invasive macroadenomas and in 75–95 % patients with microadenomas.9–11
Medical treatment is considered an adjuvant therapy in patients experiencing residual disease after surgical removal of the tumour. The only situations in which medical treatment with somatostatin analogues (SSAs) have been considered as a first-line therapy is in cases of macroadenomas with no local mass effect and a poor chance of surgical cure because of an extrasellar extension, for example in the cavernous sinus, in patients who refuse surgery and in patients with an important surgical risk. Radiotherapy is recommended for patients with acromegaly with unsuccessful surgical and/or medical therapy. The disadvantages of radiotherapy include the time delay between radiation administration and disease remission and the risk of hypopituitarism. Medical treatment is commonly admitted as an adjuvant therapy in cases of residual disease after surgery.5
The three classes of drugs available for treatment of acromegaly are SSAs, dopamine agonists and GH-receptor antagonists. SSAs were introduced in the 1980s, with octreotide being the first SSA to become available for clinical use.
Lanreotide was the second SSA introduced for clinical use and was developed in the 1990s. The first available pharmaceutical form of lanreotide was relatively short acting and required multiple daily dosing or subcutaneous infusion and this was soon superseded by the long-acting formulations of lanreotide.12 A long-acting preparation of sustained release (SR) lanreotide extended the time interval of intramuscular injection to 7–14 days.13,14
There are several forms of octreotide available for medical therapies that are listed in Table 1.
Other medical treatments are available in acromegaly and include dopamine agonists, such as bromocripitine or cabergoline. The advantage of this class of treatment in comparison to octreotide is their oral administration and lower cost. However, their biochemical effects seem to be lower in comparison with octreotide.15–17 Pegvisomant is a GH receptor antagonist that is administered subcutaneously and has been used in the US since 2003 and in Europe since 2004. The longterm efficacy of this treatment is interesting as a monotherapy with reported normalisation of IGF-1 in 89 to 97 of cases.18,19
In the recent ACROSTUDY, which included 1,288 patients in real-life clinical practice, 88 % of patients received once per day pegvisomant alone, while 36 % were administered daily pegvisomant with another treatment. The results showed a biochemical control in 63 % patients after 5 years.20 Two smaller studies showed a better biochemical response ranging from 84 % to 88 % in patients receiving a more optimal dose titration.21,22
Combination therapy including octreotide and one of the other of treatments seems to be a valuable therapeutic option in some cases. Several studies have evaluated the different combination options. Concerning the addition of dopamine agonists to SSAs a meta-analysis including 15 studies showed that adding dopamine in previously uncontrolled patients permit a normalisation of IGF-1 in 50 % patients.23 Addition of pegvisomant in patients on octreotide therapy for those with uncontrolled acromegaly leads to a normalisation of IGF-1 in about 95 % of patients.23,24
Mechanisms of Action of SSAs
The use of long-acting release (LAR) octreotide for treatment of acromegaly is supported by more than 20 years of experience and clinical research.25 This synthetic somatostatin binds to somatotstatin-receptor subtypes 2, 3 and 5 and inhibits the release of 5-hydroxytryptamine (5-HT) and the secretion of IGF-1, insulin, glucagon, secretin, gastrin, pancreatic polypeptide (PP), vasoactive intestinal peptide (VIP) growth hormone and motilin.26 Octreotide acts through four mechanisms to decrease abnormal GH secretion in acromegaly. First, octreotide suppresses GH secretion from the pituitary gland and from GH-secreting adenomas. Second, it decreases GH binding to hepatocytes. Third, it inhibits hepatic IFG-1 production and finally it controls tumour growth.27
SSAs Form and Schema of Administration
The available SSAs are octreotide and lanreotide. Octreotide is available for subcutaneous and intramuscular administration. The LAR of octreotide is delivered with an intramuscular injection in polymeric microspheres.28
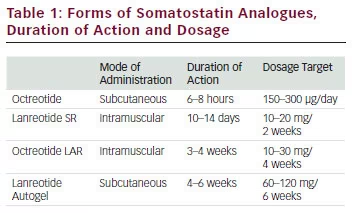
The starting dose proposed for octreotide LAR is 20 mg every 4 weeks that can be progressively increased to 30 mg every week and rarely to 40 mg every week when there is resistance.29,30 Experts recommend a trial of octreotide 2 weeks before beginning LAR octreotide. For subcutaneous (SC) octreotide, the initial dose proposed is 50 μg injected SC three to four times per day with a maximal dose of 1.5 mg per day. Lanreotide is another long-acting SSA and is available in one form: lanreotide Autogel (ATG). Currently, lanreotide ATG is the only formulation available in the US. Lanreotide ATG is an aqueous solution that is administered subcutaneously and is available in 60, 90 and 120 mg mixtures.
Lanreotide ATG has a more favourable pharmacokinetic profile than SR lanreotide, which permits deep SC administration once every 28–56 days and by self-injection rather than muscular injection every 7–14 days.31 Elimination of octreotide is principally via biliary excretion. However the pharmacokinetic profile of octreotide is only slightly altered in patients presenting moderate to severe hepatic insufficiency, requiring no adjustment of its dosage.32 Clinicians should be cautious in patients already taking cyclosporin because of a potential decreased resorption of this drug by octreotide.33 Drugs that are metabolised by cytochrome P450 3A4, such as quinidine, have a reduction in their clearance in patients taking octreotide. Concerning octreotide itself, no drugs have been reported to alter its clearance.
Efficacy of SSAs
The efficacy of octreotide treatment as a primary treatment among different studies has shown that it achieves biochemical remission in a substantial number of patients.34–36 The rate of biochemical response varies from 34 % to 60 %. The rate of significant tumour shrinkage (>20 %) was estimated in about 75 % of patients included in a prospective study.34 An important retrospective study based on the German Acromegaly Register compared primary therapy with octreotide in 145 patients to surgical treatment in 554 patients. The rate of IGF-1 and GH normalisation was higher in the group of patients treated with surgery further supporting the role of tumour surgical resection as the mainstay in the treatment of acromegaly. 37
Different studies have assessed octreotide as a neoadjuvant therapy before neurosurgery. Notably, tumour size reduction was observed in about 35 % of patients in the preoperative period and the biochemical remission rate after surgery was between 24 % to 42 % for patients first treated with octreotide before surgery versus 10 % to 23 % for patients who only benefited from surgery.38–40
A randomised, placebo-controlled study including 108 patients with acromegaly receiving lanreotide ATG 60, 90 or 120 mg every 28 days
has shown that after four injections in half of the patients achieved biochemical remission.41 A systematic review has showed a total of 33 % of patients experienced tumour shrinkage during lanreotide SR or ATG treatment in varying degrees from 10 to 77 %.42 A meta-analysis of 44 studies involving 1,852 patients compared the efficacy of lanreotide SR and octreotide LAR as postsurgical therapies. The results showed that the biochemical efficacy of LAR octreotide is greater than that of lanreotide SR among patients unselected for prior SSA responsiveness. However, there are some limitations in the results of this meta-analysis because the biochemical criteria for acromegaly remission were heterogeneous among the studies.43 When comparing the efficacy of LAR octreotide versus lanreotide ATG there was no significant difference in biochemical control or in tumour shrinkage.44,45 Lanreotide has the advantage of a prolonged injection interval in 50 % of patients.46
New somatostatin receptor ligands (SRLs) are being evaluated in clinical trials. A next-generation SRL – pasireotide (SOM230) – has a 40-fold greater affinity for somatostatin receptor 5 gene (SSTR5) than octreotide. A randomised, multicentre, open-label, crossover study including 60 patients has showed a biochemical response of 27 % after 3 months of pasireotide. Another study with nine patients receiving pasireotide during 24 months showed a biochemical control of 33.3 %.47,48 This molecule may be more effective than lanreotide and octreotide for biochemical control and can be considered as in interesting future treatment with potential advantages over currently used SRLs. The chimeric molecule dopastatin was used in phase II trials and had poor results causing its development to be stopped.
Side Effects of SSAs
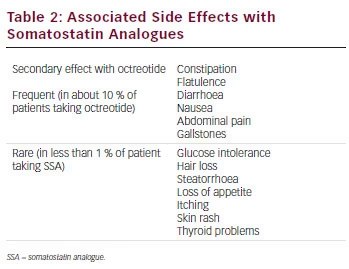
SSA therapy is generally well tolerated in most patients and treatment discontinuation related to adverse effects (AEs) is mainly due to transient gastrointestinal symptoms 49–51 (see Table 2). The most frequent side effects of SSAs are diarrhoea, abdominal discomfort, nausea and flatulence in relation to the inhibition of pancreatic exocrine secretions. In rare cases, development of malabsorption and steatorrhoea can occur and the introduction of pancreatic enzymes can resolve the problem. By inhibiting cholecystokinin secretion, ocretotide causes the formation of a more lithogenic and viscous bile leading to gallbladder sludge and gallstones in up to 30 % to 60 % of the patients, respectively.52 Ursodeoxycholic acid and chenodeoxycholic acid can be considered as a valid option to dissolve gallbladder sludge. Erythema, injection-site discomfort and burning sensations are also frequently reported by patients under SSA treatment. These side effects can be avoided by warming the drug to room temperature before injection (storage of octreotide is required in a refrigerator). The formation of painful subcutaneous nodules following repeated administration of lanreotide ATG has been described.53
SSAs can also reduce gastrin secretion with a direct inhibitory effect on the secretion of intrinsic factor increasing the risk of chronic gastritis and pernicious anaemia. Surveillance should be carried out for pernicious anaemia and a B12 injection should be considered in such patients.
Concerning the potential effects on glucose homeostasis, octreotide influences remain difficult to predict. Indeed, octreotide may induce both inhibition of insulin secretion, but also improve insulin sensibility by inhibiting GH that itself cause insulin resistance.
A meta-analysis of 619 patients treated with octreotide showed no effects on glycated haemoglobin (HbA1c) or fasting glucose levels, but showed a worsening response to OGTT.54 It could be considered that ocreotide can induce diabetes and patients treated with this therapy with previous diabetes may encounter reduced or increased insulin requirements and thus a regular control of blood glucose is recommended when SSA treatment is started. On the contrary, patients may also experience episodes of hypoglycaemia because of the inhibition of glucagon secretion. Hypothyroidism is another possible complication caused by octreotide because of the possible suppression of thyroid-stimulating hormone.
Others side effects that must be explained to patients are hair loss and bradycardia. Diffuse alopoecia remains a rare complication of octreotide therapy that occurs less frequently with the slow-release preparation and is fully reversible after discontinuation.
Clinicians should also be aware of possible kidney injury induced by octreotide as a case of acute kidney failure caused by octreotide therapy for acromegaly has been reported.55
Concerning pregnancy, data about octreotide use remain scarce as treatment was discontinued in most cases during pregnancy. There are, however, few studies showing that pregnancy is occasionally associated with a symptomatic increase of GH secretion during pregnancy. Medical treatment during pregnancy with dopamine agonists or SSAs appears to not cause significant complications or teratogenicity, but it may be associated with altered neonatal weight. Nevertheless, gestation may have a positive impact on acromegaly control both during and following pregnancy.56–58 In the absence of consistent data about octreotide, clinicians should weigh out the potential risks against the possible benefits of octreotide treatment during pregnancy.
Finally, no tachyphylaxia has been reported during long-term therapy for patients taking octreotide for acromegaly; however, a tachyphylaxia was observed in a patient taking octreotide for a neuroendocrine gastrointestinal tumour.
Conclusion
SSAs are considered a mainstay in acromegaly therapy. Its efficacy is clearly demonstrated by numerous studies. However, clinicians and patients should be aware of the possible AEs, which can often be easily controlled and rarely require the discontinuation of treatment.


