Recent advances have bolstered the anticancer therapeutic armamentarium. However, despite the enhanced efficacy and better survival offered by these newer agents, side effects remain a source of concern.1 Gastrointestinal side effects, organ toxicities and dyselectrolytaemias are critical adverse consequences. Among these, dyselectrolytaemias are multifactorial and need to be monitored and managed appropriately.1 In this review, we aim to describe the pathophysiology and abnormalities of sodium homeostasis occurring as a consequence of anticancer medications.
Literature search strategy
The medical literature for this review was identified through PubMed searches for articles published from inception to March 2020, by use of the terms ‘hyponatraemia’, ‘hypernatraemia’, ‘sodium abnormalities’, ‘sodium homeostasis’, ‘dyselectrolytaemia’ in combination with ‘antineoplastic agent’, ‘anticancer agent’, ‘cancer chemotherapy’; ‘vinca alkaloids’ including ‘vincristine’, ‘vinblastine’, ‘vinflunine’; ‘platinum containing compounds’ including ‘cisplatin’, ‘carboplatin’, ‘oxaliplatin’; ‘alkylating agents’ including ‘chlorambucil’, ‘cyclophosphamide’, ‘ifosfamide’, ‘busulphan’, ‘melphalan’; ‘immune checkpoint inhibitor’ including ‘ipilimumab’, ‘nivolumab’, ‘pembrolizumab’, ‘atezolizumab’, ‘avelumab’, ‘durmalumab’; ‘monoclonal antibodies’ including ‘cetuximab’, ‘panitumumab’, ‘alemtuzumab’, ‘trastuzumab’, ‘ado-trastuzamab emtansine’, ‘rituximab’, ‘crelizumab’, ‘obinutuzumab’, ‘veltuzumab’, ‘ofatumumab’, ‘bevacizumab’; ‘immunomodulators for cancer’ including ‘cytokines’, ‘interferon-α’, ‘interleukin-2’, ‘thalidomide analogues’, ‘thalidomide’, ‘lenalidomide’, ‘pomalidomide’, ‘chimeric antigen receptor T cell therapy’, ‘axicabtagene ciloleucel’, ‘tisagenlecleucel’; ‘tyrosine kinase inhibitors’ including ‘imatinib’, ‘dasatinib’, ‘nilotinib’, ‘bosutinib’, ‘axitinib’, ‘sorafenib’, ‘brivanib’, ‘gefitinib’, ‘erlotinib’, ‘afatinib’; ‘mammalian target of rapamycin inhibitors’ including ‘temsirolimus’, ‘everolimus’; ‘proteasome inhibitors’ including ‘bortezomib’, ‘carfilzomib’, ‘ixazomib’; ‘histone deacetylase inhibitor’ including ‘vorinostat’, ‘romidepsin’, ‘belinostat’; ‘hormonal therapy for cancer’ including ‘goserelin’, ‘leuprorelin’, ‘leuprolide’, ‘triptorelin’; ‘ancillary therapy for cancer’, ‘opioids’ including ‘codeine’, ‘morphine’, ‘apomorphine’, ‘hydrocodone’; ‘non-steroidal anti-inflammatory drugs’ including ‘ibuprofen’, ‘indomethacin’; ‘tricyclic antidepressants’ including ‘amitryptiline’; ‘anticonvulsants’, including ‘pregabalin’, ‘gabapentin’, ‘carbamazepine’; ‘hypouricosuric agents’ including ‘allopurinol’, ‘febuxostat’, ‘rasburicase’; ‘proton pump inhibitors’, including ‘omeprazole’, ‘esomeprazole’, ‘pantoprazole’; ‘osteoporosis therapy’ including ‘zoledronic acid’. Relevant articles were also identified through Google Scholar searches. Articles identified from these searches and related references cited in those articles were reviewed. Only articles published in English language were included.
Physiology of sodium homeostasis
Sodium homeostasis is closely linked to plasma osmolality. Usually, plasma osmolality is maintained between 275–290 mOsmol/kg and serum sodium between 135–145 mmol/L, with variations depending on the assays used. Sodium balance is intricately regulated by the concerted action of different neurohumoral systems.2–5
Change in plasma osmolality
Hypothalamic osmoreceptors function as the primary defence against a rising plasma osmolality and stimulate thirst by activating neurons projecting to the hypothalamic supraoptic and paraventricular nuclei.6 On the other hand, a decrease in plasma osmolality is countered by suppression of antidiuretic hormone (ADH) or arginine vasopressin (AVP) secretion.7 AVP is usually secreted above a plasma osmolality threshold of 280–290 mOsmol/kg and acts on V2 vasopressin receptors (V2R) located in the renal collecting tubule and distal convoluted tubule to increase permeability and reabsorption of water.8,9
Effective arterial volume
The homeostatic mechanisms that maintain effective arterial volume also play an important role in sodium homeostasis. The changes in effective arterial volume are sensed by the arterial baroreceptors (carotid sinus and aortic arch), the atrial volume receptors and the juxtaglomerular apparatus in the kidney.10–12 A fall in blood pressure enhances the sympathetic activity to enhance cardiac output and induce vasoconstriction, and vice versa.13 In response to volume or pressure overload, atrial receptors control the release of atrial natriuretic peptide from the atria and brain natriuretic peptide from the ventricles. Both these peptides have diuretic, natriuretic and vasodilatory properties.14 Hypotension induced by a substantial reduction in effective arterial volume can stimulate the non-osmotic release of AVP.15
Renal blood flow and sodium delivery
The juxtaglomerular apparatus secretes renin in response to a reduction in renal blood flow or a decrease in sodium delivery to the distal convoluted tubule, and activates the renin-angiotensin-aldosterone system (RAAS). Unlike baroreceptor mediated acute changes, activation of the RAAS causes sodium and water retention with resultant elevation in arterial tone and blood pressure, in a more sustained manner.
Sodium homeostasis in patients with cancer
Sodium imbalance is common in patients who have cancer.17–20 Hyponatraemia is more commonly observed in comparison with hypernatraemia.20 The derangements can be categorised as follows.
Hyponatraemia
Hyponatraemia has a heterogenous aetiology including iatrogenic causes, and occurs with an incidence that ranges between 4–44%.17,21 Syndrome of inappropriate secretion of ADH (SIADH) is the most common cause and can occur as a paraneoplastic manifestation, as a response to central nervous system (CNS) lesions (intracranial tumours, meningitis, cranial irradiation, primary or metastatic lesions), pulmonary disease (pneumonia, tuberculosis, metastasis) or as an adverse effect of cancer therapy.19,22–24 Conversely, certain drugs like cyclophosphamide (CYC), nonsteroidal anti-inflammatory drugs and antiepileptic drugs (e.g., carbamazepine) can increase renal ADH sensitivity, causing a nephrogenic syndrome of inappropriate antidiuresis (NSIAD).25
Apart from SIADH, usual causes of hypovolaemic (e.g., due to renal or extra-renal fluid losses) and hypervolaemic (e.g., heart failure, cirrhosis) hyponatraemia can also occur in individuals with cancer.26 Renal salt-wasting syndrome (RSWS) can occur due to cytotoxic therapy, adrenal insufficiency or very rarely from paraneoplastic atrial natriuretic peptide or brain natriuretic peptide secretion.26–28 Glucocorticoid deficiency, hypothyroidism, or both, can result from hypopituitarism due to tumours affecting the sellar region and can cause hyponatraemia.29 Surgical therapies such as ileal or jejunal duct choleresis after biliary drainage, and prostatic or uterine irrigation during interventions, can also cause hyponatraemia. Cerebral salt-wasting syndrome (CSWS), a close differential diagnosis of SIADH, can occur after surgery for pituitary tumours, acoustic neuromas or gliomas, and is accompanied by hypovolaemia, a feature that is not associated with SIADH.30 Thus, hyponatraemia can occur as a consequence of cancer or its complications, and as a result of anticancer or ancillary therapy. Quantifying the contribution of specific chemotherapeutic agents in the pathogenesis of hyponatraemia remains a challenge.
Pseudohyponatraemia
Falsely low sodium measurements can occur in individuals with multiple myeloma, Waldenström’s macroglobulinaemia and malignant lymphoproliferative disorders (due to hyperglobulinaemia), as well as in those with severe hypertriglyceridaemia or hyperglycaemia.31,32 These abnormalities need to be considered when interpreting reports for certain patients with cancer.
Hypernatraemia
Hypernatraemia occurs less often than hyponatraemia and has multifactorial aetiologies (including fluid restriction, diuretic therapy, glucocorticoid administration and renal concentrating defects) due to tubular damage induced by drugs such as platinum compounds, ifosfamide, amphotericin, cidofovir and foscarnet. Furthermore, diarrhoea, vomiting and insensible losses from perspiration that occur in neutropenic fevers can contribute to hypovolaemic hypernatraemia. Rarely, diabetes insipidus can cause hypernatraemia, but only when fluid intake is restricted.20
Overview of anticancer drugs
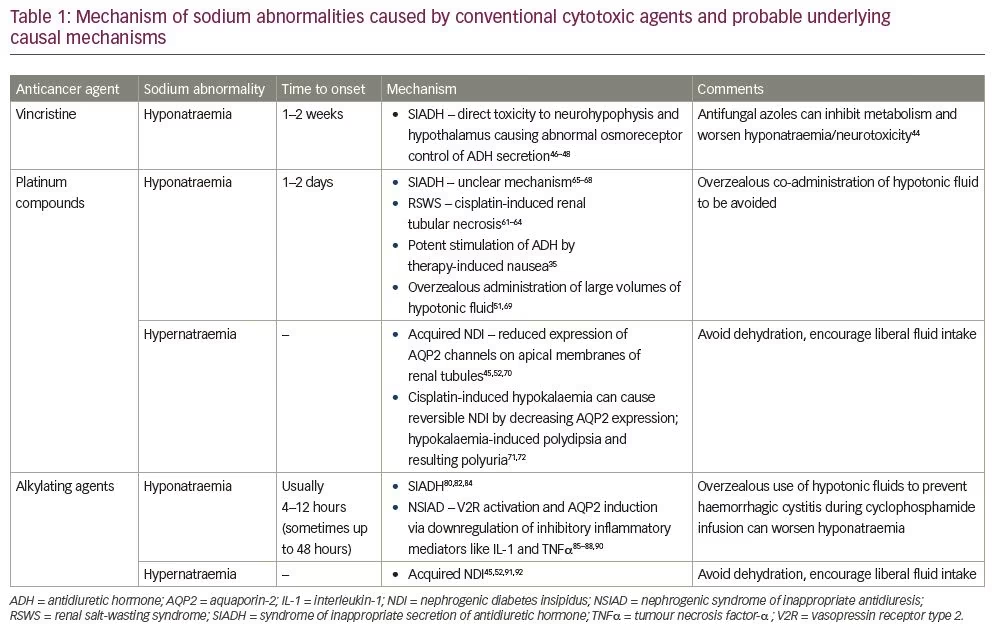
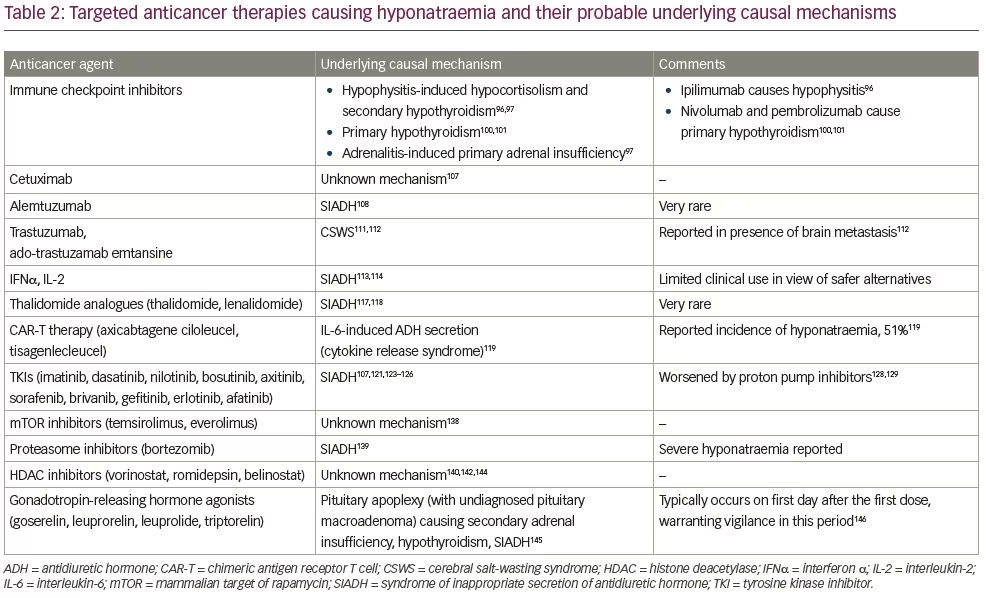
The advent of ‘personalised medicine’ has revolutionised anticancer strategies over the last two decades. While time-tested conventional systemic chemotherapy is still widely in use, there is a growing interest in the development of newer agents that ‘target’ specific processes or proteins in cancer cells that differentiate them from normal healthy cells. Targeted therapy is different from conventional chemotherapeutic agents, which indiscriminately affect all cells (healthy and cancerous), causing more adverse effects.33 Conventional agents include antimetabolites, alkylating agents, antimicrotubular agents, antitumour antibiotics and topoisomerase inhibitors, amongst others. Targeted therapy encompasses monoclonal antibodies (mAbs), including immune checkpoint inhibitors, some immunomodulators, hormonal agents and small molecule inhibitors. Small molecule inhibitors comprise tyrosine kinase inhibitors (TKIs), mammalian target of rapamycin (mTOR) inhibitors, proteasome inhibitors, matrix metalloproteinases and heat shock inhibitors, and several other investigational agents.34 Tables 1 and 2 summarise the effect of conventional anticancer agents and targeted therapies on sodium homeostasis, respectively.
Vinca alkaloids
Among vinca alkaloids, vincristine and, less commonly, vinblastine can cause hyponatraemia.35–37 The incidence of vincristine-related suspected or proven SIADH in a paediatric series of patients with acute lymphoblastic leukaemia varied between 5.9% (5/84) in a Polish series, 11.2% (68/605) in a study from the USA and 10.8% (18/166) in a report from India.38–40 A history of hyponatraemia correlated with the occurrence of neurologic complications and was found to be a marker of overt CNS leukaemia.38 The newer agent, vinflunine, can also cause hyponatraemia, with an incidence of approximately 12% (6/51).41 Severe hyponatraemia (defined as serum sodium <130 mmol/L in the study) was found in 11.9% (10/84) of children on chemotherapy for acute lymphoblastic leukaemia and occurred shortly after administration of vincristine.38 Acute life-threatening hyponatraemia with seizure and coma has also been reported with vincristine.42 In a retrospective analysis, Asians were found to be over-represented among patients developing vincristine-induced hyponatraemia, though the possible reason behind this observation is not apparent.43
Given with antifungal azoles (itraconazole, posaconazole, voriconazole and ketoconazole), the incidence of SIADH with vincristine is as high as 44% (21/47). Antifungal agents inhibit vincristine metabolism, which can lead to an increase in drug levels and subsequent neurotoxicity.44
Hyponatraemia commonly occurs after a delay of 1–2 weeks following administration of vincristine and lasts for about 2 weeks. It is often preceded by neurological manifestations that are not directly related to hyponatraemia, such as paralytic ileus or paresthaesia.45 Doses of vincristine ranging from 1.2–2.0 mg/m2 have been reported to result in hyponatraemia.35 In two patients, one with metastatic malignant melanoma and the other with a primitive neuroendocrine tumour, a lower risk of hyponatraemia was observed when vinblastine (0.4 mg/kg) was given on days 1 and 4, instead of consecutively on the first 2 days of the regimen.37
Underlying causal mechanism
SIADH is usually responsible for the development of hyponatraemia and presumably occurs as a result of the direct toxic effect on the neurohypophysis and hypothalamus, leading to abnormalities in the osmoreceptor control of ADH secretion. The presence of abnormally elevated serum and urine ADH levels that has been observed concomitantly with clinical hyponatraemia in several patients corroborates this hypothesis.46–48 Further, the neurotoxic effects of vincristine have been histopathologically demonstrated in animal models (chicks and rats), as well as in human studies.49–51 The autopsy report of a patient who had received vincristine showed the presence of axonal spheroids in the ansa lenticularis and the area surrounded by the substantia innominata, amygdala and supraoptic nucleus. It was proposed that these spheroids interfered with the inhibitor function of the supraoptic nucleus. Peripheral neuropathy is also presumed to occur from direct neurotoxicity of vincristine.51
Occurrence of hypernatraemia has not been attributed to vinca alkaloids. Vinblastine can cause nephrogenic diabetes insipidus (NDI); however, hypernatraemia has not been reported in connection to NDI.52
Platinum-containing compounds
Platinum-containing anticancer drugs include cisplatin, carboplatin and oxaliplatin. They can cause multiple dyselectrolytaemias, affecting magnesium, potassium, calcium and sodium balance.53 Hyponatraemia and, rarely, hypernatraemia are the commonly reported sodium abnormalities.
Hyponatraemia
The incidence of hyponatraemia after intravenous administration of cisplatin was 67.2% (317/472) in a retrospective evaluation over 5 years in a middle-aged population with mixed cancer types. In that report, hyponatraemia was mild in 56.6% (267/472) of recipients (categorised as 130–137 mmol/L in the study); 8.9% (42/472) had serum sodium between 120–129 mmol/L; and only 1.7% (8/472) had sodium levels below 120 mmol/L. The median time for progression of serum sodium levels to below 129 mmol/L was 7 days.54
A significant rise in urinary N-acetyl-β-glucosaminidase levels within the first 2 days after cisplatin infusion is reported to be a predictor of cisplatin-associated severe hyponatraemia.55 Other important risk factors for hyponatraemia induced by cisplatin are old age (>65 years), presence of small cell lung cancer or oesophageal cancer and low sodium levels (<138 mmol/L) at the onset of therapy.54 Carboplatin, oxaliplatin (used for advanced gastric cancer) and nedaplatin (used for untreated advanced or relapsed squamous cell lung carcinoma) have a lower incidence of hyponatraemia as compared with cisplatin.35,56–58
Underlying causal mechanism
There is evidence supporting the contribution of RSWS as well as SIADH in the pathogenesis of hyponatraemia.58–60
Renal salt-wasting syndrome
Cisplatin-induced tubular necrosis causes magnesium, potassium and calcium loss, and possibly renal sodium loss causing hyponatraemia, corroborating the possible role of RSWS.61,62 A direct relationship between the dose and occurrence of RSWS has not been clearly defined in the literature. It can happen a few days to several months after medication exposure, suggestive of a cumulative renal effect. Recovery can occur in days to months, or it may persist.63,64
Syndrome of inappropriate secretion of antidiuretic hormone
Literature suggests that SIADH could also play a part in the development of hyponatraemia in some cases. The clinical characteristics and temporal profile are distinct from that of RSWS. Onset is early, occurring in the first 2 days after administration of cisplatin, and sodium levels normalise rapidly after removal of the offending drug.65–68
Other causes
Additional underlying mechanisms that may contribute to hyponatraemia include stimulation of ADH by therapy-induced nausea and overzealous co-administration of large volumes of hypotonic fluid to prevent nephrotoxicity.35,51,69
Hypernatraemia
Both cisplatin and carboplatin can cause acquired NDI and hypernatraemia, resulting from reduced expression or impaired delivery of aquaporin-2 (AQP2) channels to the apical membrane of renal tubules.45,52,70 Hypokalaemia from cisplatin can also lead to the development of reversible NDI by decreasing the expression of AQP2 in the distal convoluted tubule.71 Recovery occurs within 1–12 weeks after the correction of serum potassium levels. In addition to NDI, direct stimulation of thirst and increased water consumption by low potassium levels also contributes to polyuria.71,72
Alkylating agents
Alkylating agents like chlorambucil, CYC and ifosfamide can cause hyponatraemia or rarely hypernatraemia.45
Hyponatraemia
CYC-induced hyponatraemia usually occurs 4–12 hours (sometimes up to 48 hours) after intravenous administration and reverses within 24 hours. It can be severe and result in seizures, lethargy or altered behaviour.73–75 It is commonly observed with high doses of CYC (30–50 mg/kg); however, it has also been reported following lower doses of 10–15 mg/kg and even after a single dose of 500 mg.73–79 In a cohort of 69 patients receiving high-dose CYC, the reported cumulative incidence of hyponatraemia (<135 mmol/L) was 52% (36), with severe hyponatraemia (defined as <120 mmol/L in the study) occurring in 5.8% (4), and symptomatic hyponatraemia in 8.7% (6).78 It has also been observed with other alkylating agents: busulfan, melphalan, glufosfamide and ifosfamide.80–82
Overzealous use of intravenous hypotonic fluids to prevent haemorrhagic cystitis during CYC infusion can aggravate hyponatraemia. An alternative approach would be to use isotonic fluids with close monitoring of sodium levels.74,75 Hyponatraemia that occurs during therapy might require discontinuation of the alkylating agent and/or fluid restriction. Use of intravenous conivaptan therapy permitted the continuation of ifosfamide along with standard hydration protocols in a patient with diffuse large B-cell lymphoma.83
Underlying causal mechanism
SIADH is the dominant mechanism behind hyponatraemia and can be due to either central release of ADH or potentiation of the renal tubular effects of ADH (NSIAD).77,83
Central syndrome of inappropriate secretion of antidiuretic hormone
The alkylating agents busulfan, melphalan, glufosfamide and ifosfamide cause hyponatraemia by increasing hypothalamic production of ADH.80,82,84
Nephrogenic syndrome of inappropriate antidiuresis
CYC causes diuresis and natriuresis in rabbits without an increase in AVP. It can activate V2R and induce AQP2 upregulation in the absence of AVP in the rat kidney.85 Intravenous CYC infusion in a patient with established central diabetes insipidus and no ability for AVP synthesis resulted in a temporary decrease in urine output with an increase in urine specific gravity, indicating a direct tubular effect of CYC or its metabolites.86 Increased interleukin-1 (IL-1) and nuclear factor-κB, a transcriptional factor of tumour necrosis factor-α (TNFα), have been shown to reduce expression of V2R and AQP2 in renal tubules.87,88 CYC-mediated suppression of these inflammatory mediators upregulate V2R and AQP2 expression in the renal tubules and has been hypothesised to induce NSIAD.89 CYC metabolites implicated in reducing the synthesis of IL-1 and TNFα in a dose-dependent fashion are mafosfamide and 4 hydroperoxycyclophosphamide.90 These observations provide a plausible mechanism of CYC-mediated NSIAD.
Hypernatraemia
Ifosfamide produces significant renal damage, particularly involving the proximal tubules and can lead to Fanconi’s syndrome. It can also cause distal tubular damage, resulting in type 1 renal tubular acidosis and NDI.45,51 CYC and bendamustine have also been associated with NDI.91,92 Hypernatraemia can occur if patients receiving these drugs have restricted fluid intake or develop dehydration from other causes, and has been reported with bendamustine.92 Hypernatraemia has not been reported as yet with CYC or ifosfamide.
Immune checkpoint inhibitors
Immune checkpoint molecules, namely cytotoxic T-lymphocyte- associated protein 4 (CTLA4) and programmed cell death protein-1 (PD-1, a cell surface receptor) and its ligand (PD-L1), prevent the immune system from destroying its cells. Immune checkpoint inhibitors are mAbs directed against CTLA4, PD-1 or PD-L1. Cancer cells usually overexpress immune checkpoint molecules to evade immune destruction and immune checkpoint inhibitors act by antagonising that effect and utilise the body’s own immune system to destroy cancer cells.93,94 The disruption of immune tolerance predisposes to immune-mediated dysfunction in endocrine glands and other organs. The relevant adverse events that can impact sodium homeostasis are hypophysitis-induced hypopituitarism, primary hypothyroidism and primary AI.94,95
Hypophysitis
Hypophysitis occurs more commonly with CTLA4 inhibitors (ipilimumab) as compared with PD-1 or PD-L1 inhibitors, with a reported frequency of around 5.6% in a meta-analysis (95% confidence interval [CI], 3.9–8.1).96 In a retrospective study comprising 154 subjects with melanoma, central hypothyroidism was reported in all 17 cases of hypophysitis, while secondary or central AI was seen in 42% (7/17) of subjects.97 Cortisol exerts a negative feedback effect on the secretion of ADH and deficiency of cortisol causes non-osmotic secretion of ADH, and that in turn causes water retention and dilutional hyponatraemia.29,98 Central diabetes insipidus secondary to immune checkpoint inhibition is unusual. There is only one reported case of avelumab-induced central diabetes insipidus secondary to infundibulo-hypophysitis, in a patient with Merkel cell carcinoma. Nocturia, polydipsia, and polyuria, which occurred 3 months after starting avelumab, reversed within 6 weeks of drug cessation.99
Primary hypothyroidism
Primary hypothyroidism is more commonly reported with PD-1 inhibitors (nivolumab, pembrolizumab) and PD-L1 inhibitors (atezolizumab, avelumab, and durmalumab) as compared with CTLA4 inhibitors.100,101 The prevalence of hypothyroidism varies from 6–20% in different series.101,102 Hyponatraemia can occur in the setting of hypothyroidism, the primary mechanism being augmented ADH release, resulting from a decrease in cardiac output-related stimulation of carotid baroreceptors.103
Autoimmune adrenalitis
Primary AI resulting from adrenalitis is an uncommon complication of immune checkpoint inhibitors. The predicted incidence of primary AI in a meta-analysis was 1.4% with ipilimumab (95% CI, 0.9–2.2), 2.0% with nivolumab (95% CI, 0.9–4.3), and 0.8% with pembrolizumab monotherapy (95% CI, 0.3–2.0). The estimated incidence from combination therapy with two different immune checkpoint inhibitors was higher (5.2–7.6%).96 In addition to the loss of inhibitory effect on ADH secretion resulting from hypocortisolism, deficiency of aldosterone further contributes to hyponatraemia by renal sodium loss and hypovolaemia-induced reflex increase in ADH secretion.104,105
Monoclonal antibodies
Epidermal growth factor receptor
Epidermal growth factor receptor (EGFR) is a transmembrane tyrosine kinase receptor that plays a critical role in the growth and survival of tumour cells. Cetuximab and panitumumab are mAbs against EGFR.106 According to the US Food and Drug Administration Adverse Event Reporting System (FAERS), there were 172 cases of acute kidney injury, 113 cases of hypokalaemia, 78 cases of hyponatraemia, 58 cases of hypomagnesaemia and 24 cases of hypertension with cetuximab.107
Alemtuzumab
Alemtuzumab is a mAb against CD52, a cell surface glycoprotein. SIADH was reported as an adverse effect of alemtuzumab in a case report.108
Trastuzumab
Trastuzumab is a mAb against human epidermal growth factor receptor 2 (HER2), approved for use in HER2-positive breast cancer and metastatic gastric cancer. Hyponatraemia has been reported in two cases with trastuzumab, but was likely caused by other concomitantly administered agents.109,110 Ado-trastuzamab emtansine is an antibody–drug conjugate comprising trastuzamab combined with the antimicrotubular maytansinoid agent, mertansine, and approved for the adjuvant treatment of HER2-positive early breast cancer. Though the initial clinical trials of this agent did not report hyponatraemia, there are rare reports of CSWS-induced hyponatraemia in patients of breast cancer with brain metastasis.111,112
Other monoclonal antibodies
Other mAbs, like the anti-CD20 antibodies (rituximab, crelizumab, obinutuzumab, veltuzumab, and ofatumumab) and the anti-vascular endothelial growth factor antibody (bevacizumab), have not been reported to alter sodium homeostasis.
Other immunomodulators
Immunomodulatory anticancer drugs of note include cytokines, such as interferon-α (IFNα) and interleukin-2 (IL-2); thalidomide analogues and chimeric antigen receptor T-cell (CAR-T) therapy, and are discussed below.
Cytokines
Cytokines such as IFNα and IL-2 have largely been replaced as anticancer agents by more efficacious and better-tolerated alternatives. There are isolated case reports of SIADH induced by these agents, presumably via a stimulatory effect on ADH secretion.113,114 Their clinical relevance is minimal, due to their limited use in routine cancer treatment.
Thalidomide analogues
Thalidomide and its analogues lenalidomide and pomalidomide mediate their anticancer effects via antiangiogenic, antiproliferative and immunomodulatory activities.115 A retrospective analysis of severe hyponatraemia among hospitalised patients reported combination therapy of thalidomide and bortezomib as a cause of development of SIADH; however, bortezomib was the more likely aetiology.116 A combination of lenalidomide and rituximab produced hyponatraemia in 9% of 30 recipients with non-Hodgkin lymphoma.117 In a phase II trial, the same combination produced hyponatraemia in 20% (9/45) of patients with non-Hodgkin lymphoma.118
Chimeric antigen receptor T-cell therapies
CAR-T therapy utilises the patient’s own modified white blood cells to kill the cancer cells. Two CAR-T therapies are currently available – axicabtagene ciloleucel and tisagenlecleucel. In a series of 78 patients receiving CAR-T therapy for diffuse large B-cell lymphoma, 51% had hyponatraemia, and 15% had sodium levels below 130 mmol/L.96,119 The mechanism of hyponatraemia is presumed to be related to IL-6-induced ADH secretion in the background of cytokine release syndrome.120
Tyrosine kinase inhibitors
TKIs competitively inhibit cellular and receptor tyrosine kinases, which phosphorylate tyrosine residues in important signal-transducing proteins. These proteins are involved in regulating cellular proliferation, differentiation, migration, metabolism and antiapoptotic signalling, and are abnormally activated in cancer cells.121,122 Hyponatraemia has been reported in a
dose-dependent fashion with the commonly used TKIs imatinib, dasatinib, nilotinib, bosutinib and axitinib.45 This has also been also observed with sorafenib (up to 39% [9/23] of cases) and brivanib (9–11%).123–125 A recent disproportionality analysis of the FAERS revealed hyponatraemia as an unexpected adverse event with gefitinib, erlotinib and afatinib.126 While it is known that SIADH causes the hyponatraemia associated with TKIs, the exact pathophysiology is unclear. It has been hypothesised that sorafenib stimulates the release of AVP by decreasing the renal papillary solute concentrations and increasing urinary osmolality.127 Proton pump inhibitors like omeprazole and rabeprazole are weak inhibitors of the cytochrome P450 3A4 enzyme. If used concomitantly with TKIs, they may increase plasma concentrations of TKI and precipitate SIADH.128,129
Mammalian target of rapamycin inhibitors
The phosphatidylinositol-3-kinase (PI3K)–Akt and the mTOR signalling pathways regulate vital cellular mechanisms controlling cell metabolism, growth, proliferation and survival.130 Abnormal activation of this pathway is related to tumourigenesis. Temsirolimus and everolimus are mTOR inhibitors approved for use as anticancer agents.130 The clinical trials of several agents of these pathways have been hindered by severe toxicities such as hyperglycaemia, dyslipidaemia, bone marrow suppression and hepatotoxicity.131 Hyponatraemia has been seen in early clinical trials of temsirolimus and everolimus.132–137 The exact mechanism of hyponatraemia is unknown.
Proteasome inhibitors
Bortezomib, carfilzomib and ixazomib are the clinically relevant proteasome inhibitors that block the ubiquitin–proteasome system, which regulates the growth of healthy and tumour cells. Severe hyponatraemia has been reported in 2.6–25.9% of patients receiving bortezomib.138 The mechanism underlying bortezomib-induced SIADH is not yet fully understood.
Histone deacetylase inhibitors
Vorinostat is an orally administered class I and II histone deacetylase (HDAC) inhibitor, which promotes cell cycle arrest and apoptosis in human haematopoietic cells and carcinoma cell lines. It is approved for use in patients with refractory cutaneous T-cell lymphoma.139 Hyponatraemia is a rare, but crucial non-haematological, dose-limiting adverse effect of vorinostat.140 Hyponatraemia has also been observed with other HDAC inhibitors like romidepsin and belinostat.141–144 There is currently no literature available regarding the risk factors or mechanism of HDAC inhibitor-induced hyponatraemia.
Hormonal therapies
Hormonal agents target the growth of hormone-dependent tumours, like prostate and breast cancer, by regulating the production or action of sex hormones. Among these agents, gonadotropin-releasing hormone agonists commonly used in prostate cancer have been associated with severe hyponatraemia, where it occurs as part of the clinical spectrum of pituitary apoplexy. Pituitary apoplexy has been reported with the use of goserelin, leuprorelin, leuprolide and triptorelin, though theoretically, it can occur with any of the agents in this class.145 With rare exceptions, almost all cases of pituitary apoplexy have ensued in the presence of undiagnosed pituitary macroadenomas, the majority of which were gonadotropinomas.145,146 Symptoms commonly occur on the first day after the first dose, with rare presentations occurring as late as the ninth or tenth day following injection.145 Patients receiving these agents should be instructed and carefully monitored regarding symptoms of pituitary apoplexy and promptly undergo evaluation in cases of any suspicion. This is particularly important for the first 10–12 days following the first dose.
Underlying mechanism
While multiple factors could contribute to the increased risk of pituitary haemorrhage and apoplexy after gonadotropin-releasing hormone agonist administration, the exact mechanism is unknown. Guerra et al. proposed a dual mechanism involving an acute and a subacute process. The acute phase is characterised by an increase in metabolic rate and local vascular perfusion due to acute release of pituitary hormones causing ischaemic changes and necrosis in an abnormally vascularised adenoma. The subacute process is believed to be due to multiple factors such as intrinsic pituitary vascular abnormalities, large size of the adenoma, elevated intrasellar pressure with rapid growth of tumour, and ischaemic change and necrosis due to compromised blood supply.145 Hyponatraemia occurs in 44% of pituitary apoplexy cases and has been postulated to correlate with hypocortisolism, hypothyroidism or SIADH, or a combination of these.147,148
Ancillary treatments
Several medications used in the supportive care setting may also cause hyponatraemia and require vigilance, especially when used alongside anticancer medications known to derange sodium homeostasis.149–159 Table 3 lists the ancillary drugs that have been found to cause hyponatraemia, along with their probable causal mechanisms.25,149–159

Management
The critical aspect of the management of hyponatraemia or hypernatraemia is to identify the aetiology. SIADH is the most common mechanism in the pathogenesis of hyponatraemia. The diagnosis of SIADH can be confirmed as per Schwartz and Bartter criteria, later updated by Ellison and Berl.160,161 The offending agent should be discontinued wherever possible. The therapeutic approach to hyponatraemia depends on its severity, rapidity of onset and symptomatology. Readers are referred to in-depth reviews by Grant et al. and Berardi et al. for detailed discussion on the management of hyponatraemia and SIADH.162,163
After administration of anticancer medicines, the onset of hyponatraemia can be within hours (e.g., with platinum-containing agents or alkylating drugs) or can be delayed by weeks (e.g., vincristine).42,54,78 Once the diagnosis of SIADH has been confirmed, discontinuation of the offending agent (if possible) should be strongly considered. Fluid restriction should be instituted in all cases of SIADH.
The decision to initiate intravenous hypertonic saline, vaptans or demeclocycline should be individualised.164 Severe acute hyponatraemia (<48 hours) is a recognised complication of CYC and will warrant administration of hypertonic (3%) saline to prevent seizures and other neurological complications.78,165 Chronic hyponatraemia (>48 hours) should be corrected slowly in order to prevent osmotic demyelination syndrome, and the rate of correction should not exceed 6–8 mmol/day.166
Levothyroxine should be started 3–5 days after starting glucocorticoid replacement, to prevent precipitation of an acute adrenal crisis in cases of immune checkpoint inhibitor-induced hypopituitarism with involvement of both axes. Administration of physiological doses of glucocorticoid usually corrects hyponatraemia, but necessitates caution as there are reports of rapid correction of chronic hyponatraemia and occurrence of osmotic demyelination syndrome.167,168 Slow up-titration of glucocorticoid doses to physiological levels in those with long-standing hyponatraemia has been suggested by some authorities to prevent this.169 Rare cases of primary AI resulting from immune checkpoint inhibitors will require mineralocorticoid supplementation, in addition to glucocorticoids.
CSWS should be managed by volume and sodium repletion, and this can be performed using a combination of isotonic saline, hypertonic saline and mineralocorticoids.30 RSWS should be similarly treated with oral or intravenous saline supplementation. Fludrocortisone has been used with varying success.63,64 Hypovolaemic or hypervolaemic hyponatraemia should be managed accordingly.
Hypernatraemia is rare, and again identification of the cause is essential for appropriate management. Slow correction of water deficit with intravenous hypotonic fluid supplementation is the mainstay of therapy.170
Conclusion
Disordered sodium homeostasis is a significant adverse effect of anticancer therapy. Hyponatraemia occurs commonly after administration of conventional anticancer agents such as vinca alkaloids, platinum compounds and CYC and, less frequently, after targeted therapy. The most common underlying causal mechanism is the induction of SIADH. Other mechanisms include primary or secondary AI, primary or secondary hypothyroidism, and increased renal sensitivity to ADH, CSWS and RSWS. Some anticancer agents have a specific temporal profile of the appearance of hyponatraemia, thus, care needs to be taken to anticipate and monitor for hyponatraemia according to the type of agent used. Certain ancillary medications, when used concomitantly, can worsen the risk of hyponatraemia, and need to be used with caution. Hypernatraemia is rare; however, it can theoretically occur in the background of drug-induced diabetes insipidus. Identification of the aetiology is central to appropriate management of an imbalance in sodium homeostasis.