Immune checkpoints are small molecules that are present on the cell surface of T lymphocytes to regulate the immune response. While some of these molecules enhance the stimulatory signals, others boost the inhibitory signals to blunt the activity of T lymphocytes (intrinsic down-regulators of immunity).1 The stimulatory checkpoints include CD28 – a family of receptors expressed on T cells – and their ligand known as B7 proteins that are expressed on the surface of tumour cells and antigen-presenting cells. The inhibitory immune checkpoints include cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), and programmed cell death protein-1 (PD-1); programmed death-ligand (PD-L)1 and PD-L2 are the ligands to the PD-1 checkpoint. Immune checkpoint inhibitors (ICIs) are monoclonal antibodies that block the inhibitory immune checkpoints, including CTLA-4, PD-1 and PD-L1, resulting in activation of T-lymphocytes to exert their antitumour immune responses.2
The ICIs that are approved by the US Food and Drug Administration (FDA) include anti-CTLA-4 (ipilimumab), anti-PD-1 (nivolumab, pembrolizumab, cemiplimab) and anti-PDL-1 (avelumab, atezolizumab and durvalumab) agents. The FDA-approved indications for ICIs include malignant melanoma, small cell lung cancer, non-small cell lung cancer, renal cell carcinoma, hepatocellular carcinoma, squamous cell carcinoma of head and neck, metastatic colorectal cancer, metastatic urothelial cancer, cervical cancer, triple-negative breast cancer, refractory Hodgkin’s lymphoma, gastric cancer, gastro-oesophageal junction cancer, primary mediastinal large B-cell lymphoma and metastatic Merkel cell carcinoma.3
As inhibitory immune checkpoints are important for immune tolerance to prevent development of autoimmune disorders, ICIs are associated with various autoimmune adverse events, known as immune-related adverse events (irAEs). Although any organ in the body can be affected, endocrine organs are commonly involved in irAEs, which are known as endocrinopathies. With increasing use of ICIs in the management of patients with advanced cancer, the incidence of endocrine adverse events is also increasing. Each ICI subgroup has varying propensity to cause a particular type of endocrinopathy and the onset time also varies. The aim of this article is to critically review the irAEs related to the use of ICIs, with special reference to the endocrinopathies, in order to enhance the knowledge of the endocrinologists, physicians and oncologists to facilitate early diagnosis and accurate treatment of these adverse effects. The currently available ICIs with their year of FDA approval, frequency of administration and immunoglobulin isotypes are shown in Table 1.
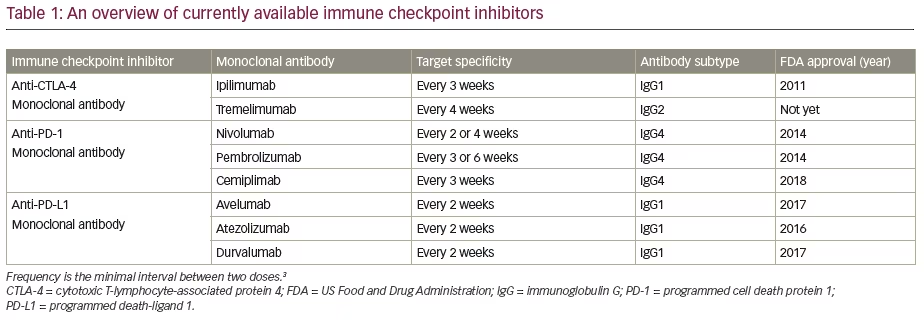
Organ-specific non-endocrine immune-related adverse events of immune checkpoint inhibitors
The irAEs of ICIs may affect almost every organ system of the body, including the gastrointestinal system, respiratory system, cardiovascular system, musculoskeletal system, nervous system, liver, kidneys, skin and blood.
Cutaneous immune-related adverse events
Cutaneous toxicity is the most common irAE, ranging from pruritus, maculopapular rash or vitiligo, to severe reactions like Stevens–Johnson syndrome and toxic epidermal necrolysis.4 Cutaneous toxicity has been reported in 45–65% of patients treated with ipilimumab, 30–40% of patients treated with anti-PD-1 inhibitors, and in 62% of cases on combination therapy with ipilimumab and anti-PD-1 inhibitors.5–8 However, severe symptoms are rare and usually do not require discontinuation of immunotherapy. Mild skin reactions, other than vitiligo, often appear within 3–6 weeks and are reversible. Vitiligo-like depigmentation has been reported in more than 25% of patients with stage III or IV advanced melanoma receiving nivolumab or pembrolizumab and is usually associated with a better response to anti-PD-1 therapy.9 Rare skin complications that may occur during combined anti-PD-1 and anti-CTLA-4 monoclonal antibody therapy include erythrodysaesthesia syndrome, urticaria, toxic epidermal necrolysis and hypersensitivity reactions to ultraviolet radiation.10
ICIs can be continued in patients experiencing grade 1 or 2 skin reactions with the use of emollients, sun protection, mild topical corticosteroids and/or antihistamines.4,7,11 In patients with grade 3 skin reactions, therapy with ICIs should be withheld, and steroid administration (prednisolone 0.5–1.0 mg/kg or intravenous methylprednisolone 0.5–1.0 mg/kg) may be needed. Restarting immunotherapy is only possible if the severity of skin lesions become grade 1 or 2. Grade 4 skin reactions mandate urgent dermatological consultation, skin biopsy and administration of methylprednisolone 1–2 mg/kg 1–2 times daily. ICIs should be permanently discontinued in such cases.4,7,11
Gastrointestinal toxicity
Gastrointestinal irAEs are the next common manifestations of ICI use and occur more often in patients receiving combination anti-CTLA-4/anti-PD-1 (44%), and single agent CTLA-4 therapy (33%) than those on single-agent anti-PD-1 therapy (20%).12 A mortality rate of 1.1% from ipilimumab-related colitis is reported.13 Severity of toxicity appears dose-dependent; for example, up to 10% with ipilimumab 10 mg/kg versus 6% with 3 mg/kg.14 Symptoms usually start 6–7 weeks after treatment initiation, with onset often later for anti-PD-1 than anti-CTLA-4, and diarrhoea is quite common. Although the descending colon is the most affected area of bowel, enteritis, oesophagitis and gastritis are also reported with occasional complications, such as toxic megacolon, ulceration, perforation and intra-abdominal abscess and perforation.12,15–17
The European Society of Medical Oncology and the American Society of Clinical Oncology guidelines recommend that mild cases be treated symptomatically with a low-fibre diet, oral rehydration and loperamide.7,18 Treatment should be initiated with oral prednisolone or budesonide for moderate cases, and intravenous methylprednisolone for more severe/non-resolving cases. Further usage of ICIs should be withheld until symptoms resolve. Infliximab is recommended in the absence of steroid response.19 There is no clinical evidence for the use of prophylactic corticosteroids to prevent gastrointestinal toxicity. Vedolizumab (α4β7 integrin inhibitor) and faecal microbiota transplant may be tried in steroid-refractory cases.20,21 Drug-induced pancreatitis is a rare complication of anti-PD-1/PD-L1 agents (<1%), although asymptomatic pancreatic enzyme elevation is much more common.22
Pulmonary immune-related adverse events
Pulmonary toxicity includes pneumonitis, and rarely, sarcoid-like reactions and pleural effusions. In a meta-analysis involving 2,716 patients, the incidence of any-grade pneumonitis was 4.6% with combination therapy and 2.1% with monotherapy.23 Mortality rates from pneumonitis are between 1% and 2%.24 The incidence of pneumonitis with PD-1 axis inhibition is higher than with that of CTLA-4. Non-small cell lung cancers and renal cell carcinomas are associated with higher risk of pneumonitis compared with melanoma, as is prior exposure to radiotherapy.25 High-resolution computed tomography (CT) is the diagnostic test for pneumonitis, and radiological patterns of acute interstitial pneumonia, cryptogenic organizing pneumonia, hypersensitivity pneumonitis or non-specific interstitial pneumonia may be seen typically involving lower lung lobes.22,25 Other investigations, such as bronco-alveolar lavage and transbronchial biopsy may be useful for diagnosis in some cases.22
Treatment depends on the severity of involvement, with subclinical radiographic changes classified as grade 1 pneumonitis (one-third of cases) requiring only treatment cessation until spontaneous resolution. Grade 2 pneumonitis, characterized by mild dyspnoea and cough, is managed with oral steroids tapered over a month on symptom resolution. Grades 3 or 4 (20–40% of cases), defined by severe and life-threatening symptoms, require hospitalization and high-dose intravenous methylprednisolone 1–4 mg/kg/day. Other agents, including infliximab, mycophenolate, cyclophosphamide and tocilizumab, may be tried in non-responders.25 Recurrence rates of 25–33% are observed on rechallenge in grade 1 or 2 disease.22,25
Hepatic adverse effects
Hepatotoxicity is frequently asymptomatic and is identified by transaminitis, which occurs nearly 6–14 weeks after receiving therapy.12 Severe hepatotoxicity is rare. Hepatitis is more frequent with dual therapy (25–30% of any grade and 15% of grade 3 or 4) than with single-agent ICI therapy (5–10% of any grade and 1–2% of grade 3 or 4).25 Lobular hepatitis, characterized by hepatocellular dysfunction is common, while ductular injury with jaundice and cholestasis is uncommon, and demonstrates poor corticosteroid response.25–29 Ultrasonography and CT scan may show steatosis, hepatomegaly, gallbladder oedema and lymphadenopathy, while liver biopsy may reveal diagnostic histological patterns.12 ICI-related hepatotoxicity requires discontinuation of the culprit drug and high-dose corticosteroids. Additionally, a second-line immunomodulatory agent, such as mycophenolate mofetil and/or tacrolimus, may be needed, and in severe cases, anti-thymocyte globulin may be used.25,30
Renal immune-related adverse events
Varying degrees of acute kidney injury with an overall incidence of 17% have been reported among patients treated with ICIs, of which, 2.2–5.0% may be related to direct insult from the drugs.31 Tubular, interstitial and glomerular disorders of different patterns are found in patients with renal involvement. Lower baseline renal function and co-administration of other nephrotoxic medications increase the risk. Although various renal imaging modalities may aid the work-up, histology is the gold-standard tool that may be necessary in some cases for diagnosis.31,32 Discontinuation of ICIs and use of steroids are indicated in moderate to severe cases.
Cardiovascular toxicity
Although myocarditis is the most common cardiotoxic feature of ICI therapy, varying degrees of pericarditis, arrhythmias, cardiomyopathy and vasculitis are also occasionally seen.33–35 Overall prevalence of myocarditis is 1.14%, which may increase to 2.4% when anti-PD-1 and anti-CTLA-4 agents are used in combination.33,34 Median time for occurrence of cardiotoxicity is 2 months, with most cases presenting within 3 months of initiation. Investigations, such as electrocardiography, cardiac enzymes and echocardiography are non-specific, and the gold-standard test for myocarditis is cardiac magnetic resonance imaging (MRI).33 Endomyocardial biopsy rarely becomes necessary. Treatment of cardiotoxicity is with discontinuation of ICIs, supportive care, corticosteroids, and less commonly, other immunosuppressants.33,35
Musculoskeletal immune-related adverse events
Various rheumatological manifestations are encountered in up to 7% of patients treated with ICIs, ranging from arthralgias, myalgias, inflammatory arthritis and polymyalgia rheumatica, to polymyositis and sicca manifestations.36,37 These features usually indicate better therapeutic response to ICIs. For grade ≥2 adverse events, including inflammatory arthritis, a referral to a rheumatologist and therapy with a tapering course of corticosteroids are necessary.38 In corticosteroid non-responders, other immunomodulatory agents may be indicated.
Neurological manifestations
Neurotoxicity is rather uncommon when compared to other toxic effects of ICI therapy. Manifestations include peripheral neuropathy, meningitis, encephalopathy, myositis and myasthenia gravis.39–41 Treatment involves discontinuation of ICIs, glucocorticoids and, occasionally, other immunosuppressants.
Ophthalmic immune-related adverse events
Ophthalmic events occur in <1% of patients, and range from scleritis, episcleritis, uveitis, retinitis, conjunctivitis, blepharitis, inflammatory orbitopathy and keratitis, to optic neuropathy, serous retinal detachment, extraocular myopathy, atypical chorioretinal lesions, immune retinopathy and neuroretinitis.42,43 Treatment involves topical steroids in mild cases, to discontinuation of ICIs with systemic steroids and immunosuppressants in severe disease.
Haemotoxicity
Commonly reported haematological manifestations of ICI toxicity are thrombocytopaenia, haemolytic anaemia and aplastic anaemia.44 Agranulocytosis and neutropenia are less often described. Management is usually successful with corticosteroids; other therapeutic strategies, such as intravenous immunoglobulins, rituximab and transfusion of blood components are rarely needed. Table 2 summarizes the organ-specific irAEs of ICI treatment.
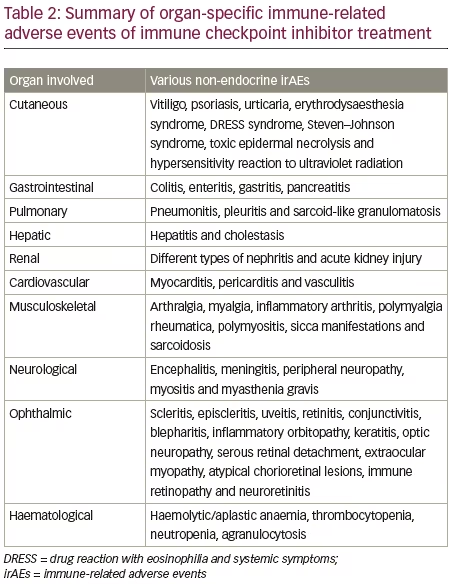
Endocrinopathies: endocrine immune-related adverse events of immune checkpoint inhibitors
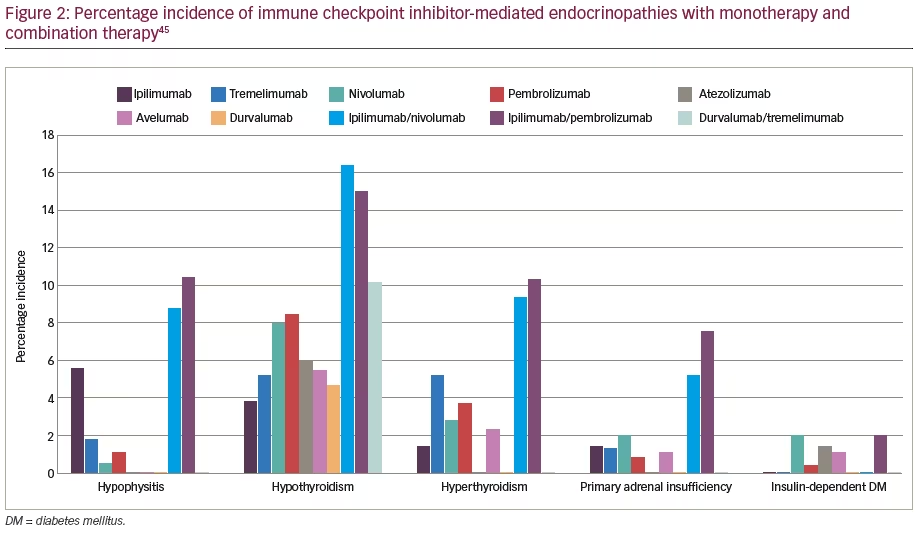
The most frequent endocrine irAE following the use of any ICI monotherapy is hypothyroidism, with hypophysitis being the next most common.45 Other endocrine irAEs, including hyperthyroidism, thyroiditis, primary adrenal insufficiency, autoimmune diabetes and hypoparathyroidism, are infrequent.45 Among patients on anti-CTLA-4 agent monotherapy, the most frequent endocrine irAE is hypophysitis, whereas among patients on anti-PD-1/PD-L1 agent monotherapy, the most frequent irAE is hypothyroidism. Patients on combination therapy with anti-CTLA-4 agents and anti-PD-1/PD-L1 agents demonstrate a higher incidence of hypothyroidism (10.2–16.4%), hyperthyroidism
(9.4–10.4%), hypophysitis (8.8–10.5%) and primary adrenal insufficiency (PAI; 5.2–7.6%). However, combination therapy rarely induced more thyroiditis (3.8–4.6%) and autoimmune diabetes (2.0%) cases compared to monotherapy.45 Fatigue is a common symptom, occurring in patients with cancer on traditional cancer therapies. However, in a patient on immunotherapy, fatigue may be a symptom of endocrinopathies like adrenal insufficiency, thyroid dysfunction and diabetes, and these symptoms mandate an endocrine work-up.
Hypophysitis
With regards to monotherapy, the incidence of hypophysitis (inflammation of the pituitary gland) is highest with the anti-CTLA-4 agent ipilimumab, with an incidence of 5.6% (95% confidence interval [CI] 3.9–8.1). This is followed by tremelimumab, which is associated with an incidence of 1.8% (95% CI 1.1–2.9).45 The disparity in incidence of hypophysitis between ipilimumab and tremelimumab is probably due to the difference in their immunoglobin subclasses (immunoglobulin G [IgG]1 versus IgG2, respectively). Anti-PD-1 agents, like nivolumab (0.5%; 95% CI 0.2–1.2) or pembrolizumab (1.1%; 95% CI 0.5–2.6), are rare causes of hypophysitis and the incidence is extremely rare with anti-PD-L1 agents.45 When used in combination therapy, the incidence of hypophysitis is 8.8–10.5%, with the incidence being 8.8% (95% CI 6.2–12.4) for ipilimumab–nivolumab, and 10.5% (95% CI 6.5–16.4) for ipilimumab–pembrolizumab combination.45
The incidence of hypophysitis is twofold greater for high-dose ipilimumab (10 mg/kg) in comparison to low-dose ipilimumab (3 mg/kg every 3 weeks for four doses).14 Moreover, the incidence was high among older age groups and males.46 Anti-CTLA-4 agent-induced hypophysitis usually develops on an average of 10.5 weeks (95% CI 9.8–11.2, p<0.001) after the onset of therapy, whereas the timing of onset of anti-PD-1/PD-L1–induced hypophysitis is extremely variable, with some cases developing in few weeks – mostly at an average of 27 weeks, and a few developing after 1 year.47 The mean time of onset for anti-PD-1 is 27 weeks (95% CI 20.9–33.1, p<0.001), and for anti-PD-L1 is 27.8 weeks (95% CI 0–58, p=0.001).47
The diagnosis of ICI-induced hypophysitis requires a high index of suspicion and is based on clinical symptoms and imaging.47 ICI-induced hypophysitis is clinically distinct from primary hypophysitis in its male preponderance, abrupt onset, older age, severe central adrenal deficiency, lower incidence of posterior pituitary and optic chiasmal involvement, and often persistent hypopituitarism. Among ICI-induced hypophysitis, anti-CTLA-4 therapy-induced hypophysitis is clinically distinct from anti-PD-1/PD-L1 therapy–induced hypophysitis in that the former is associated with symptoms of mass effect (headache or visual-field defects), panhypopituitarism, and significant changes on MRI with diffusely enlarged pituitary, widened pituitary stalk or altered contrast enhancement. In contrast, anti-PD-1/PD-L1 therapy–induced hypophysitis is associated with isolated severe adrenocorticotropic hormone (ACTH) deficiency without any symptoms of mass effect or MRI changes.47
The presenting symptom of anti-PD-1/PD-L1–induced hypophysitis is that of hypoadrenalism, whereas anti-CTLA-4–induced hypophysitis presents with hypothyroidism, hypogonadism, headache and visual disturbances.47 However, at the time of diagnosis of hypophysitis, nearly equal numbers of patients in either group (97% in anti-PD-1/PD-L1 agent versus 95% in anti-CTLA-4 agent) exhibit central adrenal insufficiency. Among the anti-CTLA-4 group, 85% manifest with central hypothyroidism and 75% with central hypogonadism. Moreover, deficiencies of growth hormone and prolactin occur in 27% of patients on an anti-PD-1/PD-L1 agent and 25% of patients on an anti-CTLA-4 agent. Diabetes insipidus is rare (2%). MRI is abnormal in 81% of cases of hypophysitis following anti-CTLA-4 agents, whereas it shows changes in only 18% of cases following anti-PD-1/PD-L1. These distinctive clinical features are important, as those patients without severe mass effect, or those not expected to have severe mass effect, should not be unnecessarily treated with high-dose steroids.47
Anti-CTLA-4 agent-induced hypophysitis can be explained by the expression of the CTLA-4 in the anterior pituitary cells that secrete ACTH, thyroid-stimulating hormone (TSH) and follicle-stimulating hormone (FSH).48 CTLA-4 expression results in the development of antibodies directed against TSH-, FSH- or ACTH-secreting cells of the pituitary gland in patients with hypophysitis who were negative for these antibodies at baseline. Moreover, CTLA-4 expression is seen in normal pituitary cells and pituitary adenomas.49 There is significant variability among individuals in the expression of CTLA-4 in the anterior pituitary cells, and this could explain why only some patients develop hypophysitis after administration of an anti-CTLA-4 agent. Anti-CTLA-4, being an IgG molecule, binds to and activates the classical complement pathway to induce an antibody-dependent cell-mediated cytotoxicity (type 2 hypersensitivity reaction), and these events further trigger autoimmune damage of the pituitary gland, resulting in hypophysitis (type 4 hypersensitivity reaction).50 The pathogenesis of anti-PD-1/PD-L1-induced hypophysitis is unknown, though the expression of PD-1 on ACTH-producing anterior pituitary cells could explain the isolated severe ACTH deficiency.47
Lymphocytic hypophysitis is characterized by infiltration of the pituitary gland by both T and B lymphocytes. However, the histopathological characteristics of ICI-related hypophysitis are not well established, as only limited numbers of biopsies are available from such cases.51 In one series, high CTLA-4 expression was associated with aggressive necrotizing hypophysitis mediated by T-cell dependent (type IV) and IgG-dependent (type II) immune mechanisms, resulting in necrosis and distortion of the anterior pituitary architecture.50
Diagnostic work-up of patients with suspected ICI-induced hypophysitis should include serum electrolytes, 8 am cortisol, ACTH, synacthen test (if indicated), TSH and free thyroxine (FT4), FSH, luteinizing hormone (LH) and oestradiol (premenopausal women) or testosterone (men), prolactin and insulin-like growth factor 1 (IGF-1).18 If symptoms of diabetes insipidus are present, evaluation with urine-specific gravity, urine and serum osmolality is indicated. MRI of the brain with dedicated pituitary sections is indicated in the presence of multiple hormone deficiencies with or without headache and visual field changes. Low cortisol with low or inappropriately normal ACTH, low FT4 with low or inappropriately normal TSH, low testosterone or oestradiol with low or inappropriately normal LH/FSH with or without hypernatraemia is diagnostic of hypophysitis.18 All patients on ICIs should be monitored for hypophysitis symptoms and, once hypophysitis is diagnosed, replacement therapy should be initiated with low-dose steroids. Central hypoadrenalism is largely irreversible, whereas gonadotrophin and TSH deficiencies are often reversible.52 Hence, initiation of levothyroxine and sex hormone replacement can be delayed. There is no significant difference between anti-CTLA-4 and anti-PD-1/PD-L1 agents as far as replacement therapy is concerned. Hypophysitis resolves rapidly once low-dose steroids are initiated and post-therapy MRI might even appear normal.46 Therefore, a normal MRI does not rule out hypophysitis. High-dose steroids should only be used in cases with severe headache and visual disturbances, as they can decrease the efficacy of ICIs, worsen the outcome and increase mortality. ICIs can be suspended/delayed until the acute symptoms of hypophysitis are resolved; however, most patients will be able to resume and continue them as previously planned.47
The standard steroid replacement is with hydrocortisone 20 mg split into three administrations: 10 mg in the morning, 5 mg at lunch time and 5 mg in the early evening. The last dose should be administered before 6 pm to avoid polyuria and insomnia. In emergency situations, intravenous hydrocortisone should be given at a dose of 100 mg followed by 50 mg every 6 hours or using continuous hydrocortisone infusion at a rate of 1–2 mg/hour. An alternative regimen to oral hydrocortisone is twice daily cortisone acetate at a dose of 25 mg in the morning and 12.5 mg in the mid-afternoon.47 Thyroid hormone replacement should be commenced several days after corticosteroid replacement is initiated, to prevent adrenal crisis. The usual dose for levothyroxine is 1.6 μg/kg/day.18 Adjustment of levothyroxine dose should be based on FT4 (not TSH), with an aim to keep FT4 at the middle or upper half of the reference range.
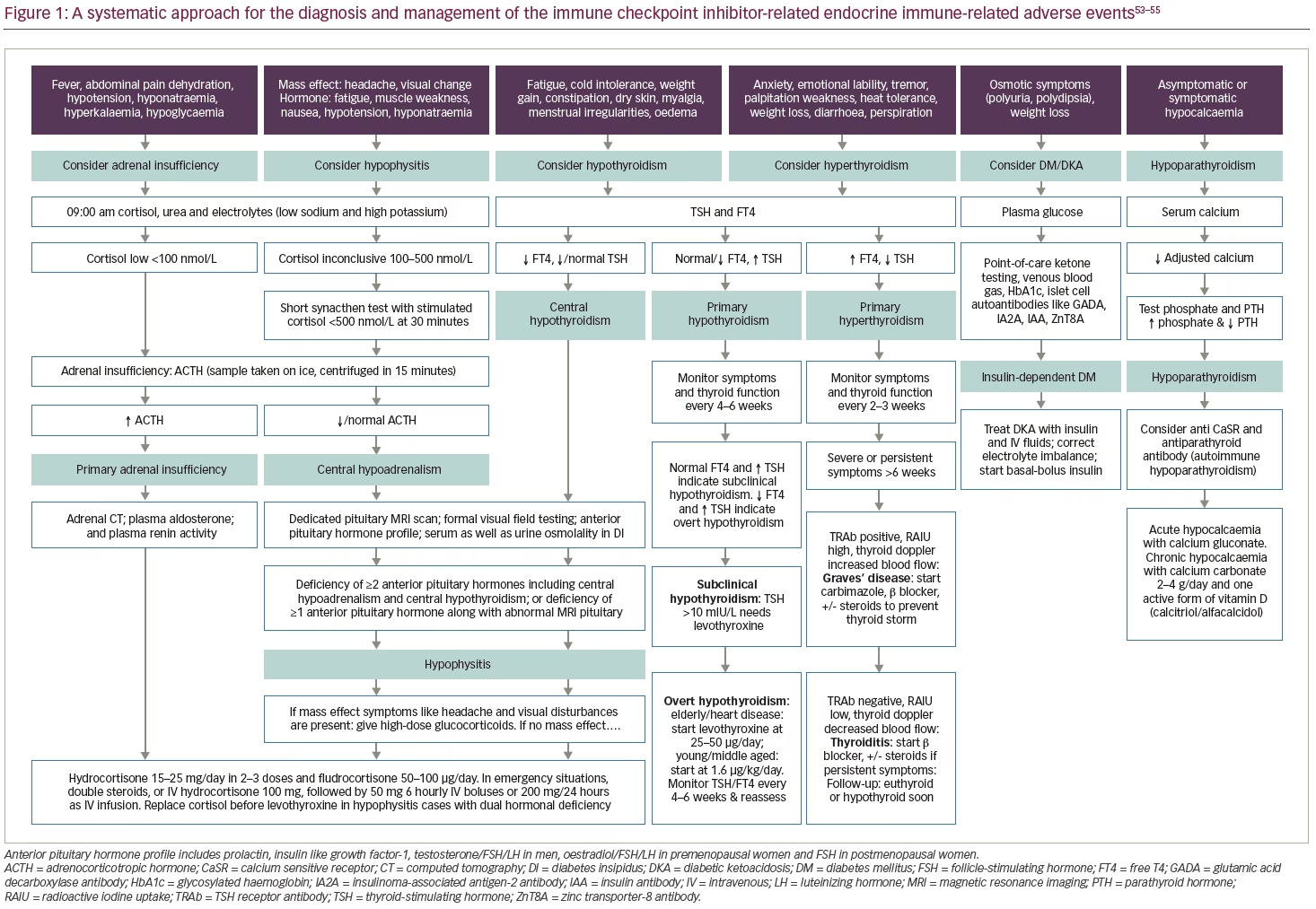
Nearly one-third of patients on ICI therapy would eventually require high-dose steroids for the treatment of non-endocrine irAEs, and long-term exogeneous steroid administration can result in hypothalamic–pituitary–adrenal (HPA) axis suppression. Hence, early-morning cortisol should not be measured to diagnose central hypoadrenalism in these patients while on high-dose corticosteroid therapy. However, early-morning cortisol can be measured once the corticosteroid dose is weaned down to approximately 5 mg prednisolone equivalent. If the cortisol level is <100 nmol/L, it strongly suggests adrenal suppression. Corticosteroid replacement should be continued and the possibility of silent hypophysitis resulting in ACTH deficiency should be considered. If the early-morning cortisol level is between 100 and 500 nmol/L, there is a possibility of adrenal suppression and hence a short synacthen test should be done. If the early-morning cortisol is >500 nmol/L, it indicates normal HPA axis and hence, the corticosteroid can be stopped.53–55
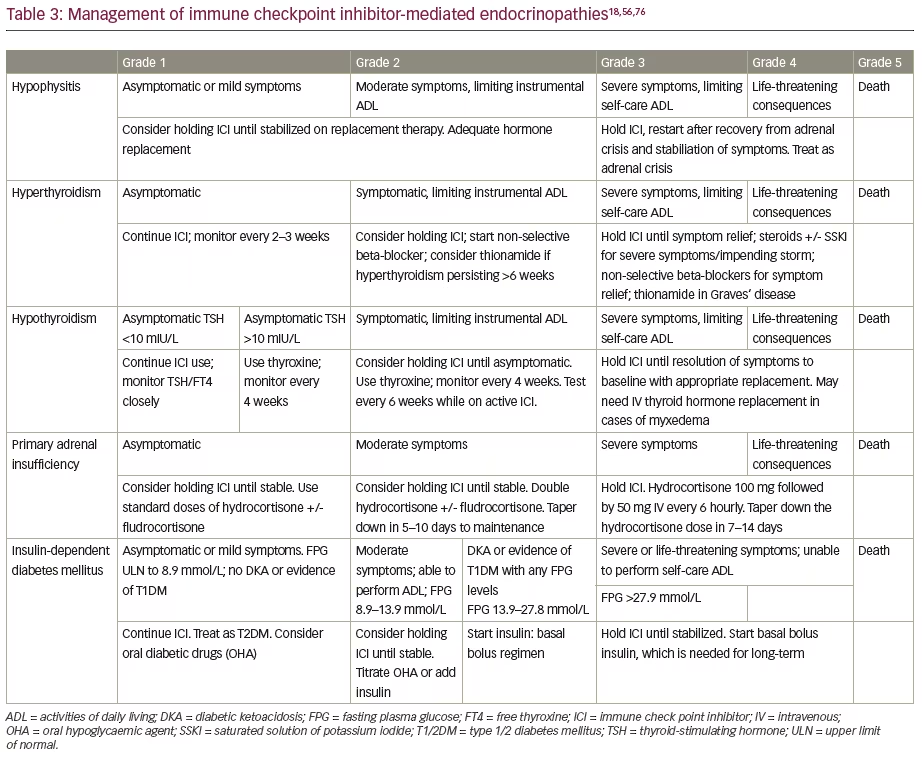
A standardized grading system proposed by the National Cancer Institute in the USA for adverse event reporting in cancer clinical trials is known as the Common Terminology Criteria for Adverse Events (CTCAE). According to the current CTCAE (version 5), ICI-induced hypophysitis can be graded into grade 1: asymptomatic or with mild symptoms; grade 2: with moderate symptoms or limitation of age-appropriate instrumental activities of daily living; grade 3: with severe, but not life-threatening symptoms or limitation of self-care activities of daily living; grade 4: with life-threatening consequences; and grade 5: resulting in death.56 The management appropriate for various grades of adverse events is given in Table 3.
Thyroid dysfunction
ICIs, especially anti-PD-1/PD-L1 monoclonal antibodies, are associated with a spectrum of primary thyroid disorders, including painless thyroiditis, overt hyperthyroidism, subclinical hyperthyroidism, subclinical hypothyroidism and overt hypothyroidism.45 Painless thyroiditis is characterized by a hyperthyroid phase followed by euthyroidism or hypothyroidism, with negative TSH receptor antibodies (TRAb), decreased uptake on radioactive iodine scan (RAIU) or decreased uptake on technetium pertechnetate (99mTc) scan and/or raised uptake on 18-fluorodeoxyglucose positron emission tomography (18FDG-PET) scan.
Hypothyroidism is the most frequent thyroid disorder related to ICI use, followed by hyperthyroidism and painless thyroiditis. ICIs may rarely cause precipitation of a thyroid storm.57 The pathogenesis of thyroid dysfunction (hyperthyroidism followed by either euthyroidism or hypothyroidism) in patients using an ICI is due to a painless destructive thyroiditis, mediated by cytotoxic T cells against the thyroid gland.58 Destructive thyroiditis develops within first few weeks of ICI therapy and a transition occurs from initial phase of hyperthyroidism to a subsequent phase of hypothyroidism within 3–6 weeks. Rarely, hypothyroidism may develop simultaneously with the onset of painless thyroiditis. The phase of hyperthyroidism is typically mild, asymptomatic and transient. Hyperthyroidism is associated with elevated thyroglobulin levels, which become normalized with the subsequent development of hypothyroidism.59
Though ICI-induced hyperthyroidism could mostly be due to destructive thyroiditis, it may rarely be due to the development of ICI-related Graves’ disease, especially with anti-CTLA-4 monoclonal antibodies.60,61 TRAb status, RAIU and colour flow Doppler ultrasonography can distinguish between destructive thyroiditis (negative TRAb, low RAIU and low blood flow on doppler) and Graves’ disease (positive TRAb, high RAIU and high blood flow on doppler). However, recent iodinated contrast media administration could suppress the RAIU in a patient with Graves’ disease. Hence, an RAIU study should be done only in patients with persistent hyperthyroidism who have not had any iodinated contrast media exposure for at least a month.62 Iodine-induced hyperthyroidism is another possible aetiology for hyperthyroidism in these patients on ICIs, who also require repeated iodinated contrast media administration for radiological investigations.63
The increased incidence of thyroid dysfunction in patients on anti-PD-1 antibodies compared with anti-PD-L1 antibodies or anti-CTLA-4 antibodies is due to the fact that the normal thyroid gland tissue expresses both PD-L1 and PD-L2 molecules, and PD-L2 blockade results in thyroid dysfunction.64 Patients with pre-existing subclinical autoimmune thyroid disease in the form of positive thyroid autoantibodies (thyroid peroxidase antibodies) are at increased risk of developing ICI-induced destructive thyroiditis and thyroid dysfunction.65
Hashimoto’s and painless destructive thyroiditis are classically associated with infiltration of the thyroid gland by both T and B lymphocytes.66 However, the histopathological features of ICI-related thyroid dysfunction are less clear as the number of biopsies available from such cases, to date, are limited. One series showed a high PD-L1 expression associated with chronic lymphocytic infiltration, granuloma formation and destruction of thyroid follicles.66 Necrotic cells, lymphocytes and histiocytes were demonstrated from biopsies in another series.67
Higher doses of ICI therapy are associated with increased incidence of hypothyroidism (2.2% versus 1.9%) and destructive thyroiditis (1.4% versus 0.6%) in patients on ipilimumab 10 mg/kg compared with 3 mg/kg.64 When used as monotherapy, the incidence of hypothyroidism is highest with pembrolizumab 8.5% (95% CI 7.5–9.7), followed by nivolumab 8.0% (95% CI 6.4–9.8), atezolizumab 6.0% (95% CI 4.2–8.4), avelumab 5.5% (95% CI 3.5–8.7), durvalumab 4.7% (95% CI 2.5–8.8) and ipilimumab 3.8% (95% CI 2.6–5.5).45 The incidence of thyroid dysfunction, including hypothyroidism, hyperthyroidism and painless thyroiditis, associated with tremelimumab, was up to 5.2% in a phase III randomized clinical trial compared with chemotherapy.68 As a combination therapy, the incidence of hypothyroidism is significantly higher compared with monotherapy, and is in the range of 10.2–16.4%. The incidence of hypothyroidism associated with ipilimumab–nivolumab combination is 16.4% (95% CI 11.7–22.5), ipilimumab–pembrolizumab combination is 15.1% (95% CI 10.6–21.8) and durvalumab–tremelimumab combination is 10.2% (95% CI 5.6–17.9).45
When used as monotherapy, the incidence of hyperthyroidism is highest with pembrolizumab 3.7% (95% CI 2.8–4.7), followed by nivolumab 2.8% (95% CI 2.1–3.8), avelumab 2.3% (95% CI 0.6–8.6) and ipilimumab 1.4% (95% CI 0.8–2.4).45 Cases of hyperthyroidism have not been reported with atezolizumab and durvalumab. The incidence of hyperthyroidism is significantly higher with combination therapy compared with monotherapy and is in the range of 9.4–10.4%, with the incidence for ipilimumab–nivolumab combination being 9.4% (95% CI 7.1–12.3)
and the incidence for ipilimumab–pembrolizumab combination being 10.4% (95% CI 6.6–16.1). Like durvalumab monotherapy, hyperthyroidism is not reported with durvalumab–tremelimumab combination as well.45
The incidence of thyroiditis is highest for pembrolizumab 2.3% (95% CI 1.2–4.6), followed by ipilimumab 2.1% (95% CI 1.1–4.1) and nivolumab 1.6% (95% CI 0.2–0.2).45 Thyroiditis has not been reported with anti-PD-L1 agents including avelumab, atezolizumab and durvalumab. Higher incidence occurs with combination: 4.6% (95% CI 2.2–9.3) with ipilimumab–pembrolizumab combination and 3.8% (95% CI 1.4–9.4) with ipilimumab–nivolumab combination.45
Clinical features during the phase of hyperthyroidism include tremor, tachycardia, fever, sweating, diarrhoea, fatigue and weight loss. Neck pain is not common in patients with destructive thyroiditis in contrast to patients with subacute thyroiditis. Periodic testing with TSH and FT4 every 4–6 weeks is advised for all patients on ICIs.18 The same tests should also be conducted in patients with symptoms of hyperthyroidism. High FT4 with suppressed TSH is diagnostic of primary hyperthyroidism. TRAb testing may be considered only if Graves’ disease is suspected (thyroid bruit or Graves’ ophthalmopathy). Close monitoring with TSH and FT4 every 2–3 weeks should be done to catch the transition to hypothyroidism that occurs with thyroiditis, or to detect persistent hyperthyroidism that is persisting for >6 weeks as in Graves’ disease.18
Management recommendations for ICI-induced hyperthyroidism are given in Table 3.18,56 Non-selective beta-blockers, such as propranolol or atenolol, should be used for symptom relief. High-dose glucocorticoids (equivalent to prednisolone 2 mg/kg/day) should only be used in patients with severe symptoms or in elderly patients with cardiovascular comorbidities. Thionamides, like carbimazole or methimazole, should only be used in patients with suspected Graves’ disease.18 Adjustment of thionamide dose should be based on FT4 measurements, with an aim to keep FT4 at the upper half of the reference range. The measurement of TSH alone is inadequate, as TSH might remain suppressed for many weeks or months after the initiation of antithyroid therapy, especially in patients with Graves’ disease. Treatment with thyroidectomy or radioiodine are options for treatment when Graves’ hyperthyroidism is not manageable with antithyroid drugs.69
Clinical features in the hypothyroidism phase include fatigue, loss of appetite, constipation, bradycardia, dry skin and weight gain.18 Periodic testing with TSH and FT4 every 4–6 weeks is advised in patients on ICIs. The same tests are used in patients with symptoms of hypothyroidism. The laboratory picture of primary hypothyroidism includes raised TSH with normal FT4 (subclinical hypothyroidism) or with low FT4 (overt hypothyroidism), while a low FT4 associated with low or inappropriately normal TSH is suggestive of central hypothyroidism associated with hypophysitis. The differentiation between primary and secondary hypothyroidism is particularly important, as thyroid replacement therapy without steroid replacement in the latter will result in precipitation of adrenal crisis. Euthyroid sick syndrome (non-thyroid illness syndrome or low T3 syndrome) is another differential diagnosis characterized by normal or low TSH, normal or low FT4, low FT3 and high reverse T3.
Management recommendations for ICI-induced hypothyroidism, based on CTCAE grades, are given in Table 3. For frail or elderly patients with multiple comorbidities including coronary artery disease, a ‘start low, go slow’ approach should be implemented, starting at a low dose of 25–50 μg/day of levothyroxine and then slowly titrating up. For people without comorbidities, the ideal body weight should be used for levothyroxine dose calculations, rather than actual body weight. The usual daily dose of levothyroxine is 1.6 μg/kg/day. Adjustment of levothyroxine dose should be based on TSH, with an aim to keep TSH in the lower half of the reference range. If adrenal dysfunction is present (which can develop as simultaneous primary hypothyroidism and PAI or as part of hypophysitis: central hypoadrenalism and central hypothyroidism), levothyroxine should be replaced several days after initiation of steroid replacement.18
Primary adrenal insufficiency
PAI is an infrequent, but severe, life-threatening adverse event following ICI therapy. It is possible that PAI is underdiagnosed, as it is often difficult to diagnose in patients who are already on steroids for the treatment of other irAEs.18 PAI is characterized by insufficient secretion of glucocorticoids with or without insufficient secretion of mineralocorticoids and androgens. PAI is associated with a rise in ACTH and/or renin. Adrenal insufficiency that occurs in patients on ICI therapy could either be central hypoadrenalism caused by hypophysitis, pituitary metastasis, or abrupt withdrawal of exogenous glucocorticoid therapy, or PAI caused by ICI-related adrenalitis, adrenal metastasis or adrenal haemorrhage. The differentiation between central hypoadrenalism and PAI is important, as the area of interest for imaging studies and the agents for treatment are different. Whereas central hypoadrenalism is treated only with hydrocortisone, patients with PAI might require a mineralocorticoid agent, fludrocortisone 100 μg/day, in addition to either hydrocortisone or prednisolone.70
When used alone, the incidence of PAI has been reported as highest for nivolumab (2.0%; 95% CI 0.9–4.3), followed by ipilimumab (1.4%; 95% CI 0.9–2.2), tremelimumab (1.3%; 95% CI 0.7–2.4), avelumab (1.1%; 95% CI 0.3–4.2) and pembrolizumab (0.8%; 95% CI 0.3–2.0). When used in combination, the incidences were much higher, in the range of 5.2% (95% CI 2.9–9.2) for ipilimumab with nivolumab and 7.6% (95% CI 1.2–36.8) for ipilimumab with pembrolizumab.45 ICI-related PAI is more common in men than women, and is common in old age (seventh decade of life). The average time of onset for PAI from the initiation of ICI treatment is 4 months.71 The pathophysiology of PAI is unclear; however, it is likely to be an autoimmune activation, as adrenal cortex autoantibodies are detected in patients with ICI-related PAI.
Patients with PAI may present with non-specific symptoms, such as nausea, fatigue, anorexia, abdominal pain, weight loss, dizziness, orthostatic hypotension; and severe PAI cases may develop features of adrenal crisis including hypoglycaemia, hyponatraemia, hyperkalaemia, refractory hypotension, muscle weakness, confusion and, infrequently, hypercalcaemia. All patients with PAI should have imaging to rule out adrenal metastasis and adrenal haemorrhage, before considering ICI-related PAI. Adrenal CT in ICI-related PAI may show evidence of adrenalitis in the form of enlargement of both adrenal glands having relatively smooth borders.72 An 18FDG-PET scan may show a transiently high uptake in the adrenal glands in patients with adrenalitis.73
Patients with a possible diagnosis of PAI should have blood taken for cortisol and ACTH before receiving hydrocortisone, if the patient is stable enough. However, blood investigations should not delay the treatment of acutely unwell patients presenting with adrenal crisis. In the setting of a low early-morning cortisol, elevated serum ACTH levels greater than twofold of the upper limit of reference range is in favour of PAI; a normal or suppressed ACTH is in favour of central hypoadrenalism. Early-morning cortisol, when used as a screening test can reduce the number of unnecessary synacthen tests by 21%, as the latter provides no additional information if the early-morning cortisol is <100 nmol/L or >500 nmol/L.74 A synacthen test will not be able to differentiate PAI from central hypoadrenalism. However, in patients with central hypoadrenalism, the synacthen test will indicate hypoadrenalism only after 3–6 weeks of any pituitary insult, including pituitary surgery, apoplexy or traumatic brain injury. This is the minimal time for adrenal atrophy to occur following ACTH deficiency.75
As PAI is associated with mineralocorticoid deficiency, measurement of aldosterone and renin is useful in its diagnosis, which is characterized by low aldosterone and elevated renin levels. If the level of aldosterone that is measured 30 minutes after synacthen injection (along with cortisol) is <5 ng/dL, it is indicative of mineralocorticoid deficiency.1,76 This test can be used in the diagnosis of an extremely rare, but possible association of PAI and hypophysitis following ICI therapy. The zona glomerulosa synthesizes aldosterone, primarily under the control of renin and plasma potassium; however, ACTH can stimulate the zona glomerulosa to increase aldosterone secretion. Moreover, the zona glomerulosa usually remains intact in central hypoadrenalism. Hence, post-synacthen test aldosterone levels show an increase in cases with central hypoadrenalism. Adrenal autoantibodies are not routinely recommended in the diagnosis of ICI-related PAI.
The management recommendations for patients with ICI-related PAI, based on CTCAE grades, are given in Table 3. All patients with PAI should be evaluated for possible triggers, including infection. Though hydrocortisone is commonly used in those with severe PAI features, intravenous use of dexamethasone 4 mg is preferred if a diagnosis of PAI is not yet established, and a short synacthen test should be conducted as soon as possible.18 All patients with a diagnosis of PAI should be given a medic alert bracelet and education regarding sick-day rules. Patients with PAI and reduced aldosterone levels, refractory orthostatic hypotension, hyponatraemia and hyperkalaemia will benefit from escalating doses of fludrocortisone.76
Immune checkpoint-related diabetes mellitus
ICI-related new-onset insulin-dependent diabetes mellitus (DM) is one of the rarer, but potentially fatal, irAEs. Most cases are secondary to anti-PD-1/PD-L1 monotherapy, though few cases are reported with anti-CTLA-4 monotherapy.45 When used as monotherapy, the highest incidence has been reported with nivolumab (2.0%; 95% CI 0.7–5.8), followed by atezolizumab (1.4%; 95% CI 0.2–9.4), avelumab (1.1%; 95% CI 0.2–7.6) and pembrolizumab (0.4%; 95% CI 0.2–1.3).45 The ipilimumab–pembrolizumab combination has an incidence of 2.0% (95% CI 0.6–5.9). ICI-related DM is more common in older men. It is characterized by a rapid onset of hyperglycaemia, rapid progression to absolute insulin deficiency and a high risk for diabetic ketoacidosis if not diagnosed and treated early with insulin. More than two-thirds of cases develop diabetic ketoacidosis as the presenting symptom. The onset of ICI-related DM occurs from 1 week to 1 year after initiation of anti-PD-1/PD-L1 therapy.45
Due to the rapid progression of hyperglycaemia, these patients have a relatively low glycated haemoglobin (HbA1c) at the time of diagnosis, with a mean level of 7.6% or 60 mmol/mol.1 Marked hyperglycaemia with normal or only minimally elevated HbA1c is a common feature in patients with ICI-related DM. The islet cell autoantibodies against glutamic acid decarboxylase, insulinoma-associated antigen-2, insulin and zinc transporter‐8 have been detected in only 50% of cases due to rapid onset beta-cell inflammation. C-peptide levels are low or undetectable in nearly all cases, indicating significant loss of endogenous insulin secretion.1 The T lymphocytes of patients with type 1 DM (T1DM) have a reduced expression of PD-1, PDL-1 and CTLA-4, implying that patients treated with ICIs are susceptible for developing T1DM. Moreover, patients with ICI-related DM shared similar human leukocyte antigen class II genes to those of patients with T1DM.1 Apart from new-onset insulin-dependent DM, anti-PD-1 monoclonal antibodies are known to worsen a pre-existing type 2 DM (T2DM).
The diagnosis and management of new-onset insulin-dependent DM use similar established principals to those used for T1DM. The diagnosis can be made during routine testing for hyperglycaemia in an asymptomatic patient on ICI therapy or the patient can present with symptoms of hyperglycaemia (polyuria, polydipsia and weight loss) or with symptoms of diabetic ketoacidosis (nausea, vomiting, abdominal pain, hyperventilation, lethargy, obtundation, seizure or coma). Patients on ICI therapy and their family members should be educated regarding symptoms or hyperglycaemia, as well as diabetic ketoacidosis. The beta-cell destruction is irreversible, hence the insulin requirement is going to be permanent in nearly all patients. Steroids have no role in the management of ICI-related DM. Management of patients with ICI-related DM are given in Table 3.
Primary hypoparathyroidism
Hypoparathyroidism is an exceedingly rare complication of ICI therapy, and there are only a handful of case reports showing an association with anti-PD-1 therapy as well as combination therapy, but not with ipilimumab monotherapy.77–79 Moreover, hypocalcaemia was observed as a common complication to anti-PD-1 treatment in a recent metaanalysis.77 The pathogenic mechanisms for hypoparathyroidism are not established. It might be related to hyperactivation of calcium-sensing receptor (CaSR) of the parathyroid glands by activating CaSR autoantibodies or mediated by an autoimmune destruction of the parathyroid gland triggered by ICI therapy. Hypoparathyroidism should be considered when hypocalcaemia is associated with low-normal or low parathyroid hormone levels. Anti-parathyroid and CaSR-activating autoantibodies may be detectable in patients with ICI-related hypothyroidism mediated by these autoantibodies. Hypoparathyroidism is largely irreversible and treatment with an activated form of vitamin D along with calcium supplementation are needed on a long-term basis.78
Hypogonadotropic hypogonadism
Transient hypogonadotropic hypogonadism can be seen in patients with hypophysitis. It will manifest as low testosterone levels (in men) or low oestradiol levels (in premenopausal women), associated with low or inappropriately normal FSH and/or LH. In post-menopausal women with typically elevated FSH and LH, inappropriately low FSH and LH indicate presence of hypogonadotropic hypogonadism. As hyperprolactinaemia is a cause of hypogonadotropic hypogonadism, prolactin levels should be checked in all these patients. However, prolactin levels are low in most cases of hypophysitis, while hyperprolactinaemia is a rare observation.1 Hypogonadotropic hypogonadism can develop in the absence of hypophysitis and is especially seen in patients on ipilimumab.80 However, it is difficult to interpret the exact aetiology of hypogonadism in this patient population, as the severity of chronic illness (advanced cancer), as well as the high dose systemic corticosteroids that are used for the treatment of various ICI-related non-endocrine irAEs, are known causes of hypogonadotropic hypogonadism.
Diabetes insipidus and syndrome of inappropriate antidiuretic hormone secretion
Diabetes insipidus is a rare endocrinopathy associated with ICI therapy, and an extremely rare feature in ICI-related hypophysitis, as this predominantly results in deficiency of anterior pituitary hormones.81 The drugs reported to cause diabetes insipidus include ipilimumab, and avelumab.82–84 The onset of diabetes insipidus occurs nearly 12 weeks after the initiation of ICI therapy in these patients.85 Another possible association, reported in a couple of case reports, is syndrome of inappropriate antidiuretic hormone secretion (SIADH), manifesting as hyponatraemia. However, both these cases had central hypoadrenalism, which might have caused hyponatraemia, and SIADH had been possibly a wrong diagnosis.86,87 Hypoadrenalism results in hyponatraemia by stimulating antidiuretic hormone secretion by various mechanisms that include direct stimulation of antidiuretic hormone release, indirect stimulation of corticotropin-releasing hormone, lowering of blood pressure and cardiac output, and upregulation of the expression of aquaporin-2 in the renal tubules.47 The resultant rise in antidiuretic hormone levels leads to free water retention and dilutional hyponatraemia.
Adrenocorticotropic hormone-dependent cortisol excess with or without Cushing’s syndrome
Transient ACTH-dependent hypercortisolaemia with or without features of Cushing’s syndrome has been reported in two case reports.88,89 Both cases developed central hypoadrenalism on follow-up, indicating the need for close monitoring to detect hypoadrenalism when ACTH elevation is detected in patients on ICI therapy. Transient ACTH elevation can be explained by a destructive hypophysitis analogous to the destructive thyroiditis.88,89
Monitoring for endocrinopathies while on immune checkpoint inhibitor therapy
Patients on ICI should be routinely monitored for the development of endocrinopathies before each cycle of ICI therapy (typically every second or third week for the majority of available agents; see Table 1 for further details), every 2–3 months for the next 6 months, and thereafter every 6 months.90 Testing should be repeated more frequently if endocrinopathy symptoms develop. Figure 1 illustrates an algorithm for the work-up and management of various ICI-related endocrinopathies. ICI-related clinically significant adrenal insufficiency manifesting with hypotension, dehydration and electrolyte disturbances, whether caused by hypophysitis (central hypoadrenalism) or by PAI, should be treated in lines of management of adrenal crisis with hospitalization, intravenous hydrocortisone and sepsis work-up. Figure 2 illustrates the percentage incidence of various endocrinopathies (endocrine irAEs) associated with the available monotherapy and combination therapy. From the figure, it is evident that anti-CTLA-4 agents, or their combination, are common causes of hypophysitis. On the other hand, anti-PD-1/PD-L1 agents, or their combination, are a common cause of thyroid dysfunction (hypothyroidism and hyperthyroidism), and the same group can rarely present with PAI and insulin-dependent DM.
Areas of uncertainty
Patients who have developed irAEs, have been seen to demonstrate superior overall survival and surrogate endpoints, like progression-free survival and recurrence-free survival, in comparison with those who have not developed them.1 Activated autoreactive T cells (decreased immune tolerance) mediate the irAEs, and people who develop irAEs have T cells that show better response to ICI therapy.1 Hence, their immune system possesses the enhanced ability to destroy the cancer cells (enhanced antitumor activity). Several retrospective observational studies support the hypothesis that endocrine,91–93 as well as non-endocrine,94–97 irAEs are associated with improved overall survival in ICI-treated patients with various types of cancers.
There is contrasting evidence regarding the severity of irAEs, overall response rate and the time to progression. One study observed improvement in overall response rate (25% versus 6%) and prolongation of median time to progression (30 weeks versus 10 weeks) in patients with high-grade (grade 3–5) irAEs compared with patients without high-grade irAEs, respectively.98 In contrast, another study observed that those with low-grade (grade 1 or 2) irAEs have higher overall response rate and prolongation of time to next therapy or death.99 The latter observation might be due to the higher morbidity associated with severe irAEs and the requirement for immunosuppressants in the management of these high-grade irAEs.100 Thus, a definite conclusion cannot be reached regarding the severity of irAEs and the overall outcome of ICI therapy until new large prospective trial data are available.
For many years it was considered that irAEs are the necessary price for the cancer immunity that ICIs provide. However, researchers are now trying to uncouple ICI-mediated antitumour activity from ICI-mediated irAEs. One way of doing this is by developing monoclonal antibodies that have dual specificity to both ligands (for example PD-L1) and immune checkpoint receptors (e.g. LAG3 or TIM-3), which are upregulated by tumour cells.101 As these monoclonal antibodies with dual specificity can be retained in the tumour microenvironment, sparing the normal tissues, they promote antitumour effects without causing irAEs.102 Another way to uncouple the antitumour activity from irAEs is by selective depletion of regulatory T cells in the tumour microenvironment with the help of pH-sensitive anti-CTLA-4 antibodies that do not result in CTLA-4 depletion in normal tissues.103
A third approach under investigation for uncoupling irAEs from the antitumour effects is known as Probody® therapeutics (CytomX Therapeutics, South San Francisco, CA, USA), which is characterized by the development of recombinant, proteolytically activated antibody prodrugs that will become active only when they get exposed to tumour-associated proteases in the tumour microenvironment.104 Conjugating ICI with matrix-binding peptides and then injecting them into or around the tumour may facilitate intra-tumoural retention with lower systemic antibody levels and lower rates of irAEs.105
Conclusions
ICI therapy is associated with endocrine and non-endocrine irAEs. Thyroid dysfunction and hypophysitis are the most common endocrinopathies, whereas PAI, insulin-dependent DM and hypoparathyroidism are the uncommon members of the group. Anti-CTLA-4 agents are more toxic compared with anti-PD-1/PD-L1 agents, as they are associated with significantly higher numbers of irAEs. Hypophysitis is more common with anti-CTLA-4 agents, whereas thyroid dysfunction, insulin-dependent DM and PAI are more common with anti-PD-1/PD-L1 agents. ICI-induced endocrinopathies are potentially fatal if not diagnosed and treated promptly. ICI-related adrenal insufficiency is mostly irreversible, and the replacement therapy often permanent. In contrast, thyroid and gonadal hormonal deficiency may be reversible. Counselling the patient and family members before ICI initiation regarding potential irAEs will facilitate early diagnosis of endocrinopathies. Finally, effective co-ordination between oncology and endocrinology departments can improve the outcome of patients with ICI-related endocrinopathies as, in contrast to other irAEs, patients with even high grades of endocrine irAEs can continue their ICI therapy, provided the hormone replacement therapy is adequate and the symptoms are controlled.