Catecholamine-secreting tumours that arise from chromaffin cells of the adrenal medulla and the sympathetic ganglia are respectively referred to as pheochromocytomas and paragangliomas. The term paraganglioma is also used for tumours derived from parasympathetic tissue in the head and neck, most of which do not produce catecholamines.1 The classic triad of symptoms in patients with pheochromocytoma consists of episodic headache, sweating and tachycardia. Approximately one-half have paroxysmal hypertension; most of the rest have either primary hypertension or normal blood pressure (BP).1 Clinicians should always consider pheochromocytoma when evaluating for secondary causes of hypertension, as missing this diagnosis can result in devastating cardiac complications, including death.1
Nowadays, genetic testing is recommended for all patients with pheochromocytoma. A germline mutation in one of the susceptibility genes identified so far explains ~40% of all cases;2 these genes include the rearranged during transfection (RET) proto-oncogene, von Hippel–Lindau (VHL)tumour-suppressor gene, neurofibromatosis type 1 tumour-suppressor gene and the genes encoding four succinate dehydrogenase (SDH) complex subunits.3 There are few cases of pheochromocytoma with TMEM127 mutation reported. We report on a patient with germ ine TMEM127 mutation-associated pheochromocytoma.
Case presentation
A previously healthy 53-year-old woman, with no past relevant medical history, had presented to her general practitioner 18 months earlier with daily pulsatile bilateral frontal headaches persisting for 6 months, reporting different characteristics than her usual migraines. She also complained of paroxysmal episodes of fatigue, palpitations, heat, sweating, and non-quantified weight gain in the previous months. Her past medical history was unremarkable, except for sporadic migraines since adolescence. She denied other relevant pathological medical history, including smoking, alcohol or drug consumption, and had no previous hospital admissions or surgeries. She was not on regular medication or supplements, including analgesic drugs or hormonal contraception. Arterial hypertension was diagnosed, no treatment started, and she underwent investigation of secondary causes of arterial hypertension. During this workup, pheochromocytoma was suspected and she was evaluated in our hospital department.
The patient reported one female paternal cousin submitted to right adrenalectomy due to a pheochromocytoma and one male paternal cousin with an adrenal incidentaloma under investigation, both followed in a different medical centre. She has two brothers, both healthy. Her mother was diagnosed with arterial hypertension at an older age and her father had no history of hypertension. The patient did not know of any other relatives with arterial hypertension.
No other relevant medical family history was reported, namely, known familial hereditary diseases.
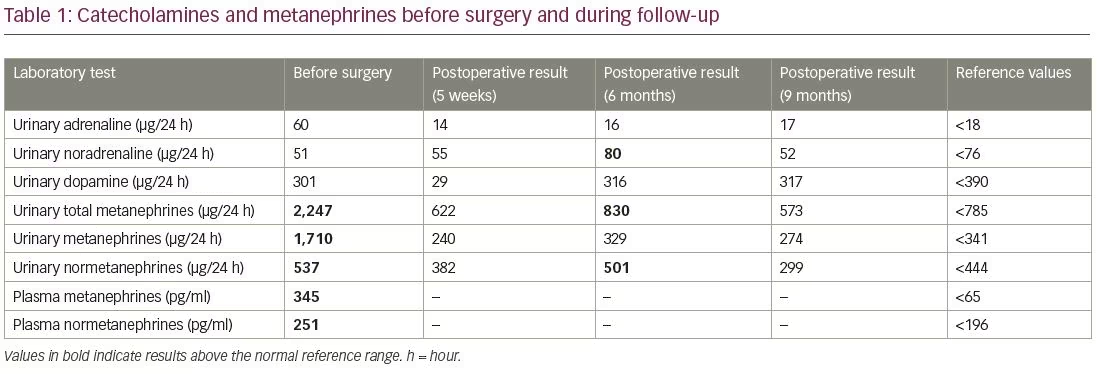
The physical examination was notable for a high diastolic BP (137/106 mmHg) and a body mass index of 25.4 kg/m2. Core blood laboratory tests including electrolytes, fasting glucose, glycated haemoglobin, hepatic enzymes, renal function and thyroid function were within the reference values. Dyslipidaemia was diagnosed (total cholesterol 256 mg/dl, low-density lipoprotein cholesterol 169 mg/dl, high-density lipoprotein cholesterol 75 mg/dl, triglycerides 97 mg/dl). Primary aldosteronism and cortisol excess were excluded; nevertheless, the patient presented elevated levels of plasma metanephrines (345 pg/ml) and normetanephrines (251 pg/ml), as well as elevated 24-hour urinary adrenaline (60 µg/24 h), metanephrines (1,710 µg/24 h) and normetanephrines (537 µg/24 h) (Table 1). Chromogranin A and neuron-specific enolase levels were normal.
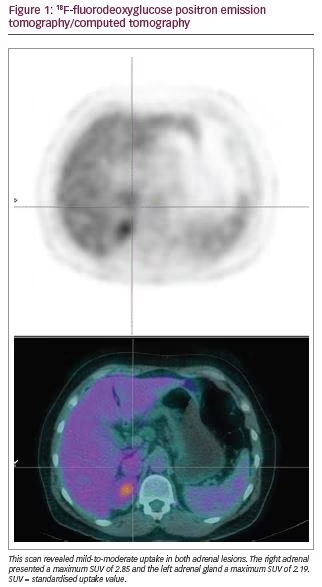
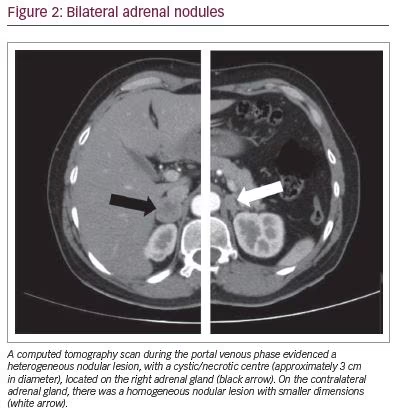
An abdominal magnetic resonance imaging scan revealed a 26 x 20 mm nodule on the right adrenal gland, a 13 x 9 mm nodule on the left adrenal gland, both described as suspicious for pheochromocytoma and a hypointense nonspecific focal image in the lower left iliac bone with 11 mm diameter. 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography (PET)/computed tomography (CT) scanning revealed mild-to-moderate 18F-FDG uptake in both adrenal lesions (right standardised uptake value [SUV] maximum: 2.85; left SUV maximum: 2.19) (Figure 1) and confirmed a hyperdense image in the iliac bone with low uptake (SUV maximum: 1.38), described as unknown aetiology, while whole body 123I-metaiodobenzylguanide scintigraphy evidenced hyperfixation only at the right adrenal gland. CT scan of the abdomen confirmed a heterogeneous nodular lesion, with a cystic/necrotic centre with nearly 3 cm diameter, located on the right adrenal gland. The contralateral adrenal gland presented a homogeneous nodular image with smaller dimensions (16 mm) (Figure 2). A 68Ga-DOTANOC PET/CT was additionally performed excluding possible neuroendocrine metastatic lesions.
Following multidisciplinary discussion, laparoscopic removal of the right adrenal gland was decided. Preoperative management was immediately initiated with an α-receptor blocker (phenoxybenzamine 20 mg/day) with normalisation of BP. Next, a β-adrenergic receptor blocker (propranolol 40 mg/day) was added. An elective laparoscopic complete right adrenalectomy was performed. Surgery was uneventful except for one hypertensive crisis during induction of general anaesthesia, controlled with sodium nitroprusside, and a hypotensive episode after clamping the vascular axis, promptly reverted with the administration of 200 µg phenylephrine. There was haemodynamic stability during the procedure, with good urinary output. Blood loss was negligible and no hypoglycaemic episodes occurred after surgery. Postoperative recovery was unremarkable and the patient was discharged after 3 days, asymptomatic, with normal BP. Gross examination of the surgical piece showed a 3 cm medullary nodule, with well-defined borders and haemorrhagic spots. Histology confirmed a pheochromocytoma confined to the right adrenal gland (pT1 Nx) with no vascular, capsular or periglandular adipose tissue invasion. No areas of necrosis were reported. Five weeks after the procedure, urinary metanephrines and normetanephrines levels had normalised. The patient remained asymptomatic with normal BP without the need for anti-hypertensive drugs.
Genetic testing became available 5 months after surgery and revealed a TMEM127 gene mutation detected in heterozygosity (NM_017849.3[TMEM127]:c.410-2A>C). Had the patient undergone a genetic analysis prior to surgery, cortex-sparing surgery would be the first choice; however, since it was available only after surgery, a total adrenalectomy had already been performed. Since there is a high risk of bilateral pheochromocytoma, surveillance was maintained with frequent clinical and biochemical evaluations. Six months after surgery, biochemical screening revealed mildly elevated levels of urinary metanephrines and normetanephrines (Table 1). An imaging reassessment (CT scan) was performed, reporting no changes on the left adrenal lesion. Nine months after surgery, urinary metanephrines and normetanephrines levels were within the reference values. The patient remained asymptomatic and is under surveillance. She was evaluated in the genetic consult and her family were offered genetic testing.
Discussion
TMEM127 is a negative regulator of mammalian target of rapamycin effector proteins, which promote cell growth and protein translation. TMEM127 protein interacts with several effector proteins of receptor kinase intracellular signalling pathways, and acts as a tumour-suppressor protein.2,4,5 While previous studies suggested a low penetrance of TMEM127 mutant alleles,6 recent data has evidenced a high penetrance of pheochromocytoma in patients with this mutation.7 Still, pheochromocytoma associated with a TMEM127 mutation is rare. In fact, Abermil et al. studied 642 unrelated patients who did not carry mutations in major pheochromocytoma susceptibility genes; 559 patients were affected by pheochromocytoma, 72 by paraganglioma and 11 by both pheochromocytoma and paraganglioma, and found a prevalence of TMEM127 mutation of 0.9%.4 Thus, the authors have suggested that TMEM127 mutation should be considered, in patients with pheochromocytoma, after a negative screening of VHL, RET, SDHB, and SDHD genes.4
An exceptional finding in patients with TMEM127 mutation-associated pheochromocytoma is the older average age at presentation, similarly to sporadic cases. A recent report suggested that the occurrence of symptoms seems to start a decade earlier than the age at diagnosis, and reported in 11 cases a mean age at diagnosis of 41.6 years,7 a decade earlier than our clinical case. The other main phenotypic association with this mutation is that the majority of reported cases presented only pheochromocytoma5,7 and rarely paraganglioma.8 Pheochromocytoma can be unilateral but frequently presents as a multicentric,7 bilateral or familial tumour.4,5 Nodular adrenomedullary hyperplasia can also be a form of presentation.7 Malignancy is rarely found among TMEM127 mutation carriers;5,9 nevertheless, TMEM127 gene mutations have been reported in rare cases of renal cell carcinoma.2 Regarding the biochemical profile, an international study that identified 19 individuals with pheochromocytoma and TMEM127 mutation reported no preference of adrenaline or noradrenaline secretion6 such as in this case; however, others have suggested a predominant adrenaline-secreting pattern.4,10,11
Due to the high prevalence of multicentric and bilateral tumours in TMEM127-related pheochromocytoma, periodic surveillance of the contralateral adrenal gland is mandatory.7 Surveillance of the surgical site of the removed adrenal in these cases is also recommended.7 This patient presented bilateral nodules, and urinary normetanephrines were slightly abnormal 6 months post-operatively, thus the patient is under tight surveillance despite a normal biochemical test at 9 months post-surgery. In order to avoid adrenalectomy with subsequent iatrogenic adrenal insufficiency, adrenal-sparing surgery should be considered in these patients.1 In this case, had the genetic test been available prior to surgery, a sparing surgery would be the preferred choice for treating this patient, highlighting the importance of genetic testing, even in older patients.
TMEM127 mutation should be sought in patients with familial, bilateral or apparently sporadic pheochromocytoma2 and older age should not discourage genetic testing. Its high penetrance of pheochromocytoma corroborates the benefits of genetic testing of at-risk relatives and mutation carriers, that should undergo clinical surveillance annually.7 This case report evidences the benefit of genetic testing for an accurate clinical management and treatment of patients and mutation carriers in TMEM127-related pheochromocytoma.