




Craniopharyngiomas (CPs) are rare, benign, epithelial tumours of the sellar and parasellar regions arising as embryonic malformations along the pathway of the craniopharyngeal duct.1 They have an incidence of 0.13 per 100,000 per year to 0.5–2.0 per million per year and are classified as grade I tumours according to the 2017 WHO Classification of Tumors of the Central Nervous System (CNS).2,3 Adamantinomatous CP (ACP) and papillary CP (PCP), are the two distinct entities of CPs with different histological and genetic patterns.4 ACP is the most prevalent subtype and displays a bimodal age distribution, between age 5–15 years and 45–60 years, whereas PCP prevalence peaks at age 40–55 years.5,6 The more challenging features of these tumours are their anatomic vicinity to vital structures (hypothalamus, optic pathways, third ventricle, circle of Willis vessels), along with their intrinsic local infiltrative tendency, which renders their growth problematic and responsible for increased rates of long-term morbidities and increased mortality compared with the general population.7,8
The current therapeutic armamentarium includes the surgery of CPs, either gross total resection (GTR) as definitive treatment, or subtotal/partial resection (STR) as a cytoreductive approach with or without radiotherapy.9,10,11 The safest approach used nowadays is STR followed by radiotherapy, unless there is a clear margin between the tumour and the surrounding vital structures, allowing a safe GTR. The intra-cystic administration of sclerosing substances, bleomycin or interferon-α, is variably used to control fluid formation and diminish cyst size.12,13 Of the two agents, bleomycin has an increased toxicity, whereas interferon-α displays an acceptable safety profile.14 Overall, the current approved management of CP is far from ideal, since a number of complications can alter the quality of life of patients with CP.10 Therefore, neoadjuvant treatment may be of great use in shrinking the initial lesion, allowing for a GTR and minimizing the postoperative complications.
On the other hand, the development of molecular-targeted therapies in oncology has increased the number of available treatments for tumours caused by genetic and molecular aberrations.15 Since no molecular-targeted or cytotoxic systemic therapy has been approved for the systemic treatment of CPs, intense research of molecular pathophysiological mechanisms has unveiled several molecular mechanisms that are implicated in CP pathophysiology. These discoveries gave rise to the introduction of medical therapy in CP management.4,16,17 The use of such drugs targeting molecular effectors of CP pathophysiology must be carefully considered, since it is expected that they will minimize the debilitating complications associated with surgery and radiotherapy, particularly in recurrent tumours, but not at the cost of serious adverse effects (AEs). The aim of this systematic review is to present the available literature on clinical studies and case reports about systemic drug therapies targeting recently identified molecular alterations in both ACPs and PCPs. The search strategy included the following Medical Subject Headings (MeSH): (adamantinous craniopharyngioma [MeSH Terms]) OR (craniopharyngioma, adamantinous [MeSH Terms]) OR (craniopharyngiomas, adamantinous [MeSH Terms]) OR (adamantinous craniopharyngiomas [MeSH Terms]) AND (papillary craniopharyngioma molecular pathology [MeSH Terms]) OR (adamantinous craniopharyngiomas [MeSH Terms]) AND (papillary craniopharyngioma medical treatment [MeSH Terms]) OR (craniopharyngioma pharmacological treatment [MeSH Terms]) OR (craniopharyngioma drug therapy [MeSH Terms]) OR (craniopharyngioma drug treatment [MeSH Terms]) AND (“2011/03/22” [Date – Entry] : “2021/03/22” [Date – Entry]) (Figure 1).
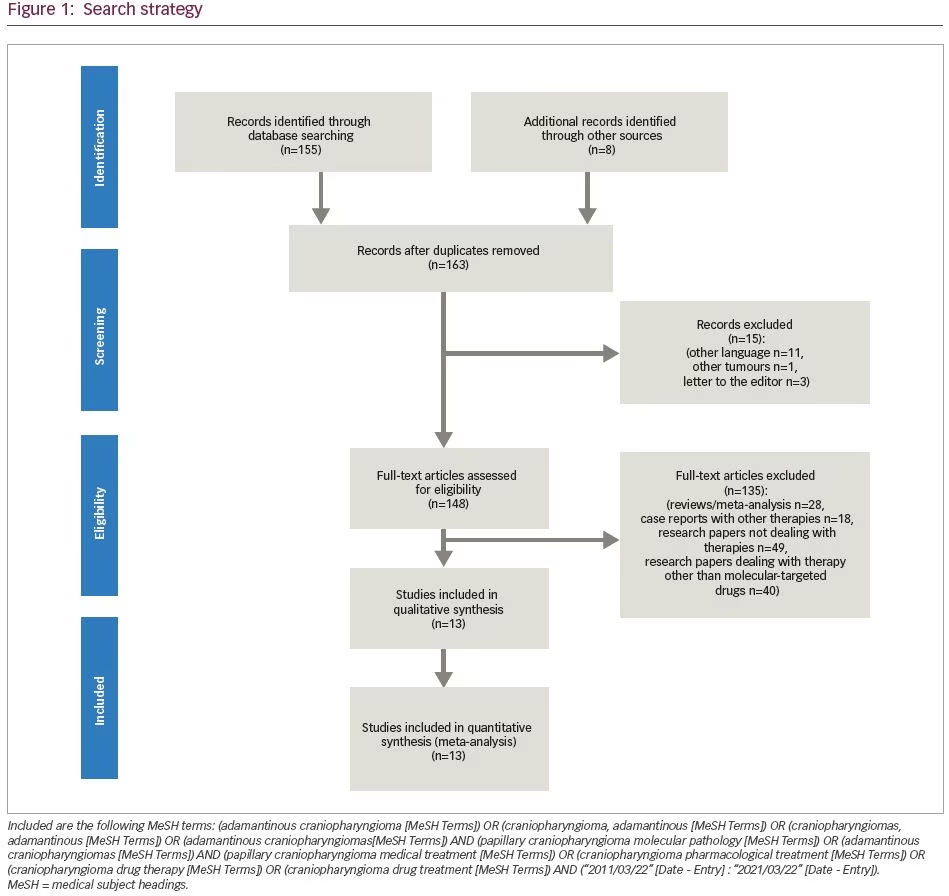
Molecular basis in medical treatment of craniopharyngiomas
Advances in molecular research have demonstrated that the two different CP types represent distinct entities, each with specific genetic and pathological features.17 In this section, we will discuss the main molecular findings that differentiate ACP and PCP, and how the biological differences have helped identify novel targeted treatments, and the use of such treatments in current clinical practice.
Molecular pathways and molecular aberrations in adamantinomatous craniopharyngiomas
Wingless pathway signalling
Activation of the canonical Wingless/Int (Wnt)/β-catenin pathway is an important pathological feature of ACP. The Wnt pathway is involved in organ formation during embryogenesis and has a pivotal role in designating and maintaining stem cell lineage.18 β-catenin, encoded by the CTNNB1 gene, is a central signalling molecule in the Wnt pathway and is involved in a variety of cell processes, including cell proliferation, differentiation and embryonic development.18,19 In the physiological state, and in the absence of WNT activation, β-catenin is marked for destruction by a complex consisting of AXIN, glycogen synthase kinase-3β (GSK-3β) and adenomatosis polyposis coli, among other proteins. In the pathological state, or following Wnt activation, Wnt binds to the N-terminal extracellular cysteine-rich domain of a Frizzled family receptor, disrupting the destruction complex of β-catenin, which is free to accumulate and translocate into the nucleus. Here, it interacts with the T-cell factor/lymphocyte enhancer factor family of transcription factor to regulate the transcription of genes implicated in cell differentiation and proliferation.20
Currently, CTNNB1 mutations are the only well-studied genetic alteration resulting in CP, and result in a degradation-resistant form of β-catenin and aberrant nuclear accumulation of the protein in certain cells within the tumour, mainly within epithelial cells located in the clusters or ‘whorls’. These cell clusters, showing nucleocytoplasmic accumulation of β-catenin, are not present in PCP or any other pituitary tumour.21
In the nucleus, β-catenin acts as a transcription factor, leading to overactivation of the canonical Wnt/ β -catenin pathway.22 These activating mutations, first described by Sekine et al.23 and subsequently found in 57–96% of patients with ACP,24,25 are located in exon 3 of CTNNB1, a key exon encoding for the serine-threonine phosphorylation sites for GSK-3β that activates degradation of β-catenin in the destruction complex (Figure 2).26 Experiments in primary human ACP cell cultures have demonstrated that increased expression of β-catenin enhances cell migration, whereas inhibiting β-catenin expression using small interfering RNA reduces cell motility and invasion capacity of ACP tumour cells.27
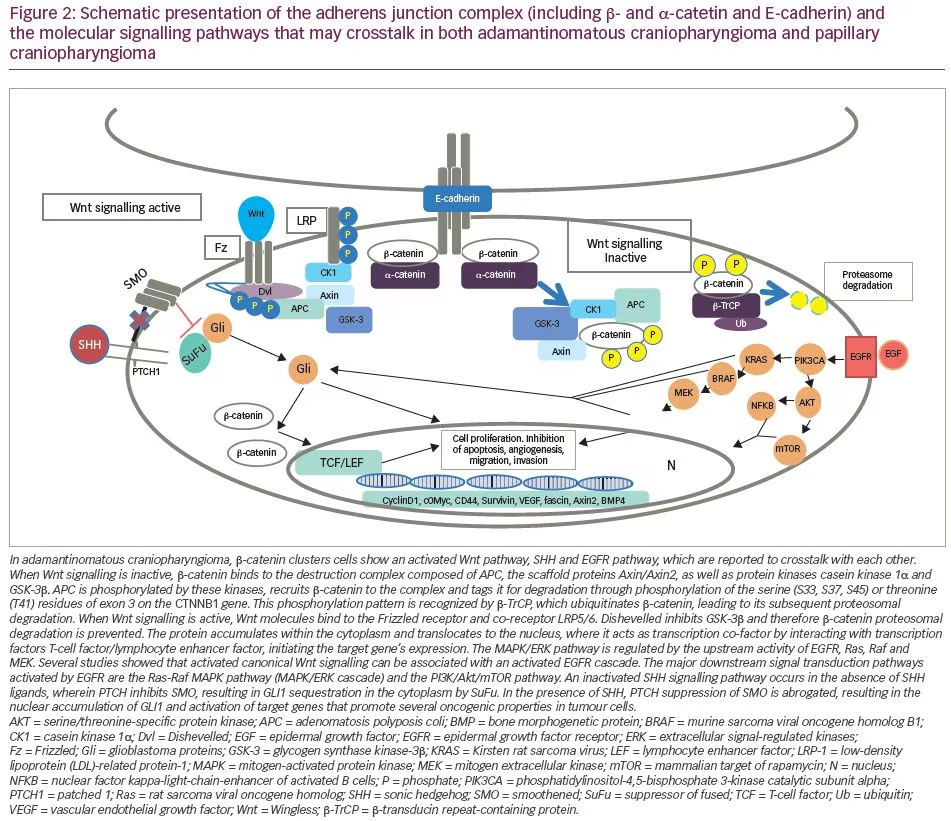
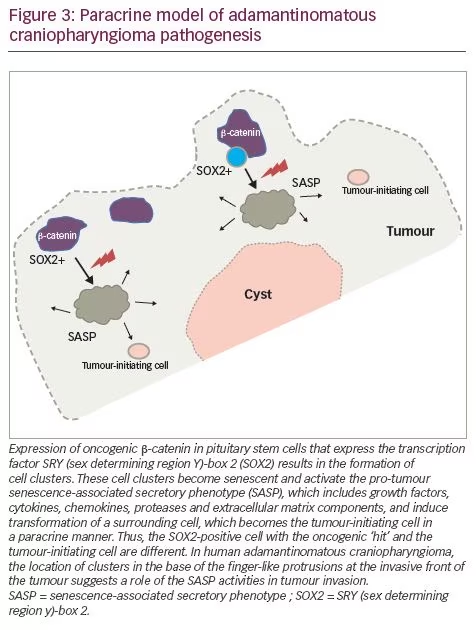
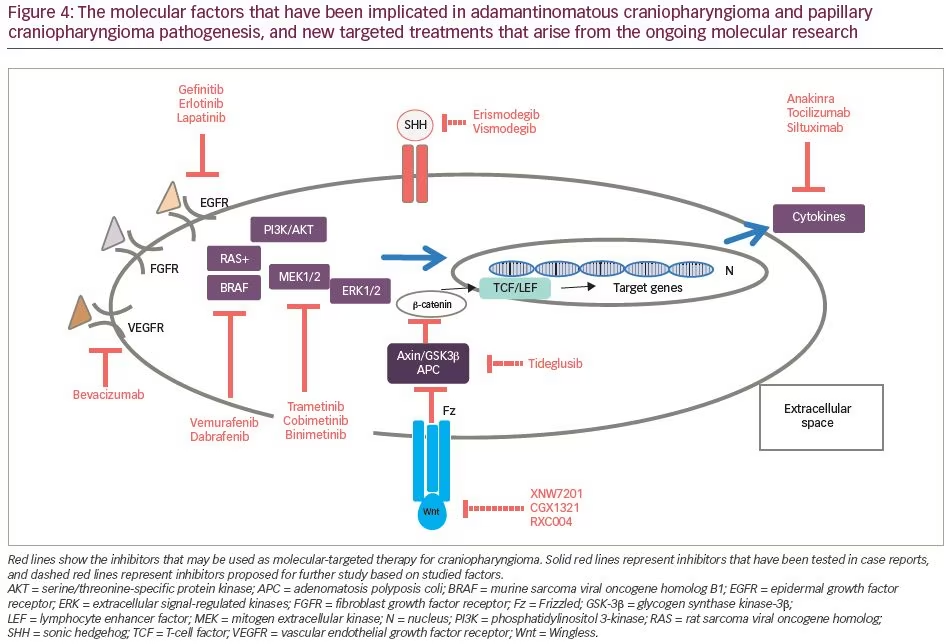
Murine studies have confirmed that CTNNB1 mutations are oncogenic drivers, capable of initiating and sustaining tumourigenesis. The expression of oncogenic β-catenin, specifically in stem cells expressing the transcription factor SRY (sex determining region Y)-box 2 (SOX2), in the murine adult pituitary results in ACP-like tumours forming in mice (Figure 3).28 Interestingly, the tumours do not derive from the SOX2-positive stem cells that express oncogenic β-catenin, but rather from SOX2-negative cells that are transformed in a paracrine manner by the SOX2-positive-derived cluster cells.29 Analyses of these murine models have revealed that β-catenin-accumulating cell clusters, as well as single cells with accumulated β-catenin, act as signalling ‘hubs’ within the tumour. These clusters secrete a plethora of growth factors and cytokines, including sonic hedgehog (SHH), cytokines (such as interleukin [IL]-1 and IL-6) and growth factors (such as epidermal growth factor [EGF], fibroblast growth factor [FGF], Wnts, transforming growth factor [TGF]-β and bone morphogenetic proteins [BMPs]), which activate specific pathways in nearby tumour cells.22,30 The role of epithelial clusters in human ACP was shown in a study by Apps et al., who used laser capture microdissection to separate cell clusters and analyse their transcriptomic profiles.30 They demonstrated high levels of the FGF, BMP and Wnt families of secreted factors, and demonstrated activation of the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathway, which was particularly prominent at the tips of the invading tumour epithelium (Figure 3).30 To test whether the MAPK/ERK pathway can be a potential therapeutic target, the authors targeted this pathway using trametinib, a specific mitogen extracellular kinase (MEK) inhibitor that has shown promising results in MAPK/ERK-driven solid tumours,31,32 and demonstrated reduced proliferation and increased apoptosis in explant cultures of human and mouse ACP (Figure 4).30
Failure to identify the CTNNB1 mutation in all ACP samples has led to speculation that other genetic mutations may underlie ACP tumourigenesis. This was further supported by the coexisting mutations in BRAF (V600E) and CTNNB1 (T41I), which were identified in two ACP tumours.33 Recently, Apps et al., using laser capture microdissection combined with tagged-amplicon deep sequencing, screened 22 ACP tumour samples: CTNNB1 mutations were identified in all samples, including those with very low mutant allelic frequencies.34 These data suggest that failure to identify CTNNB1 mutations in a low proportion of ACP tumours may be due to the lower sensitivity of the sequencing technology used in previous studies.
While Wnt pathway/β-catenin inhibitors could successfully target the Wnt signalling cascade, there have been concerns regarding their safety profile, as they may also affect the normal Wnt-dependent activities.17 Therefore, direct targeting of Wnt pathway/β-catenin is not currently considered among intervention strategies (Figure 4).35
Inflammation
A classic feature of ACPs is the presence of cysts, which are routinely larger than the solid portion of the tumour and can exert significant mass effect on adjacent structures.1,5 Intra-cystic therapies alone or in combination with surgery are a recommended treatment approach to prevent cyst expansion and pressure to the surrounding anatomical structures.
Cytometric bead analysis of cyst fluid and transcriptomic data of the solid tumour compartment have revealed elevated concentrations of cytokines and chemokines, including IL-6, IL-8, IL-10 and C-X-C motif chemokine ligand 1 (CXCL1), in fluid from ACP tumours compared with fluids from other paediatric brain tumours and healthy brain tissue.36 Cyst fluid characterization has also revealed elevated concentrations of indoleamine 2,3-dioxygenase 1 (IDO-1), an immunosuppressant, and the presence of β-thymosins, peptides with anti-apoptotic and anti-inflammatory properties.36,37
The human monoclonal antibodies tocilizumab, an anti-IL-6 receptor, and siltuximab, an IL-6 inhibitor, approved by the US Food and Drug Administration (FDA) for the treatment of inflammatory diseases such as juvenile idiopathic arthritis, Castleman disease and cytokine release syndrome, could be potential treatments for cystic CP. In two cases with recurrent, cystic ACP, tocilizumab alone or in combination with bevacizumab resulted in a significant cyst shrinkage.38 Anakinra, an IL-1 receptor inhibitor, may also be of use by preventing inflammatory pathway activation (Figure 4).30
The mitogen-activated protein kinase/extracellular signal-regulated kinase and epidermal growth factor receptor pathways
The MAPK/ERK and EGF receptor (EGFR) pathways are two additional signalling pathways involved in the growth and development of ACPs and therefore could be alternative potential therapeutic targets. In healthy cells, the MAPK/ERK pathway, in response to extracellular stimuli, mediates cellular growth, proliferation and survival.39 Abnormal MAPK signalling may lead to increased or uncontrolled cell proliferation and resistance to apoptosis, and has been implicated in many different types of cancers.40 As previously mentioned, Apps et al. established that the MAPK/ERK pathway is dysregulated in human and mouse ACP tumours; they showed high levels of several members of the FGF, TGF-β and BMP families of secreted factors, which signal to neighbouring cells, as evidenced by immunostaining against the phosphorylated proteins pERK1/2, pSMAD3 and pSMAD1/5/9 in both human and mouse ACP.30
In 2011, Hölsken et al. identified that activated EGFR in ACP cells significantly promoted tumour cell migration, which was inhibited using the tyrosine kinase inhibitor (TKI) gefitinib in vitro.26 Although the authors did not identify any mutations or amplifications in the EGFR gene, EGFR phosphorylation was detectable in tumour cell clusters at the brain infiltration border, and co-localized with nuclear β-catenin and Fascin, indicating that activating EGFR may have a role in brain infiltration.26
The ACP transcriptome of 15 ACP samples revealed several pharmaceutical targets that were significantly and consistently overexpressed in these tumour samples. Among the most highly expressed were: several targets of the kinase inhibitor dasatinib (lymphocyte-specific protein tyrosine kinase [LCK], ephrin type-A2 [EPHA2] and SRC); EGFR pathway targets (epiregulin –[AREG], EGFR and receptor tyrosine-protein kinase erbB-3 [ERBB3]), which were inhibited by drugs such as cetuximab, erlotinib, lapatinib; and other potential cancer targets, such as SHH, Matrix metallopeptidase 9 (MMP9) and 12 (MMP12), which were inhibited by erismodegib and vismodegib (SHH) or AZD1236 (MMP9, MMP12).41 EGFR signalling has also been proposed as an escape mechanism for radiation therapy leading to radioresistance in ACP through enhanced survivin expression.41,42 Inhibition of EGFR activity by the TKIs gefitinib or CUDC-101 acts as a radiosensitizer on ACP tumour cells, leading to increased cell death (Figure 4).41
Cellular senescence
Molecular profiling and immunohistochemistry analyses have revealed that the cluster cells in both mouse and human ACPs contain senescent cells. Senescence is a cellular programme that induces a stable growth arrest mediated by the p53/p21CIP1 and p16INK4A/pRb tumour suppressor pathways.43 Senescence typically occurs in response to damaging stimuli, including telomere shortening (replicative senescence), DNA damage (DNA damage-induced senescence) and oncogenic signalling (oncogene-induced senescence).43 Even though senescent cells are unable to re-enter the cell cycle, these cells are metabolically highly active and secrete a plethora of factors, including proinflammatory cytokines, chemokines, growth factors and proteases, referred to as the senescence-associated secretory phenotype (SASP) (Figure 3).44 The SASP recruits immune cells to senescent cells, thereby facilitating their elimination, which serves a tumour suppressor function. Paradoxically, however, the SASP has been shown to promote tumour cell progression through secretion of factors that promote angiogenesis, extracellular matrix remodelling or epithelial-mesenchymal transition (EMT).44 The role of cellular senescence has already been investigated in other brain benign neoplasms, such as pituitary adenomas.45
One study has shown that mouse and human ACP oncogenic β-catenin clusters contain senescent cells with an activated SASP, and that attenuating the senescent/SASP response in murine cluster cells, either genetically or in aged mice, significantly reduces tumour-inducing potential.46 This study was the first to point at senescence and the SASP as causative agents of non-cell-autonomous tumourigenesis in vivo and to explain the tendency of human ACP to relapse even after radiotherapy, as activating a senescent phenotype in human clusters could make them resistant to conventional anticancer therapies, such as radiotherapy.47 The development of senolytic agents,48 and the association of components of the SASP with cellular programmes such as EMT, indicate that further investigation of senescence in the context of ACP is warranted.
Programmed cell death
The inhibition of immune checkpoints, in particular cytotoxic T-lymphocyte antigen-4 and programmed cell death protein-1 (PD-1) and its ligand, programmed cell death-ligand 1 (PD-L1), has evolved as a promising and effective treatment against a growing number of malignancies, and its discovery as a potential cancer therapy was the subject of the 2018 Nobel Prize in Physiology.49,50 Recently, there have been some reports of a possible role of PD-1 and PD-L1 in the pathogenesis of ACP; Coy et al. identified increased expression of PD-1 in nuclear β-catenin accumulation whorl-like tumour cell clusters, whereas PD-L1 was found in a significant number of tumour and immune cells of the cyst lining.51 The consistent co-occurrence of nuclear β-catenin and PD-1 expression with concomitant elevated downstream MAPK and mammalian target of rapamycin (mTOR) signalling may implicate β-catenin transcriptional activity, indicating that therapeutic targeting of PD-1 may have intrinsic therapeutic efficacy independent of any interaction with the immune system.51 The presence of PD-L1 in the cyst lining indicates a possible interaction with the highly proinflammatory cytokine pattern identified in the ACP cyst fluid as previously described. The strong PD-L1 expression in cyst-lining cells may represent an immune checkpoint barrier that modulates the activity of immune cells within the cyst fluid. Furthermore, and considering that increased PD-L1 expression correlated with elevated mTOR and MAPK signalling in ACP, inhibiting PD-L1 may also affect the EGFR and MAPK/ERK pathways and inhibit tumour growth.51
The sonic hedgehog pathway
The SHH signalling pathway is an important pathway expressed early in embryogenesis, and acts as a major regulator of cell differentiation, cell proliferation and tissue polarity; therefore, it has an essential role in organogenesis, including Rathke’s pouch.52 SHH has been implicated in different types of cancer tumourigenesis, as well as in tumour progression and metastasis.53 Upregulation of the SHH pathway in both mouse and human ACP has been described by several investigators.22,41,54 While inhibiting the SHH pathway significantly reduced basal cell carcinoma progression in several cases,55 it has proven detrimental in other cancers, such as pancreatic ductal adenocarcinoma, where enhanced tumour progression and aggressiveness was observed in both preclinical and clinical trials.56 Recently, Carreno et al.57 showed that treating ACP with vismodegib, an FDA-approved SHH pathway inhibitor, results in a significant reduction in median survival in murine models, due to premature development of highly proliferative and vascularized undifferentiated tumours. Reinforcing these mouse data, inhibiting the SHH pathway in human ACP leads to a significant increase in tumour cell proliferation both ex vivo, in explant cultures, and in vivo, in a patient-derived xenograft model, indicating that vismodegib, and potentially other smoothened (SMO) inhibitors, may be contraindicated in patients with ACP.57
Molecular pathways and molecular aberrations in papillary craniopharyngiomas
Until recently, the genetic event underlying the development of PCP had remained unclear. Several studies employing whole exome sequencing, next-generation panel sequencing, pyrosequencing and Sanger sequencing have demonstrated the presence of activating mutations in BRAF (V600E) in PCPs, with a reported prevalence of 81–100%, depending on the sequencing method used.16,33,54,58 The expression of the mutant protein is confirmed by immunohistochemistry using an anti-BRAF-V600E antibody.59 Although this mutation is predicted to result in the activation of the MAPK/ERK pathway in all tumour cells, immunohistochemistry against pERK1/2 – read-outs of the active MAPK/ERK pathway that promote cell growth, proliferation, migration and survival60 – has revealed that only a small proportion of epithelial cells lining the fibrovascular cores express pERK1/2 despite the expression of BRAF-V600E throughout the tumour.59
It is interesting that despite the broad expression of the BRAF-V600E mutant protein in the tumours, pERK1/2 staining is mostly restricted to cells surrounding the fibrovascular cores. These pERK1/2-positive cells were shown to express the pituitary stem/progenitor markers SOX2 and SOX9, suggesting that these lining cells may represent tumour stem cells.59 Double immunofluorescence staining revealed the co-expression of SOX2/SOX9+ and Ki67 in basal cells around the fibrovascular cores, implying that proliferating SOX2-positive cells may be driving PCP growth.59
Targeting the molecular pathways of craniopharyngiomas involved in the current clinical setting
Systemic medical therapy in adamantinomatous craniopharyngiomas
Summarizing the aforementioned molecular pathways, activating mutations of the Wnt pathway gene CTNNB1, driven by a single recurrent somatic mutation of CTNNB1,23 can be recognized in most ACPs. However, the activation of the Wntβ-catenin pathway has not yet translated into novel molecular-targeted treatments. In a smaller fraction of ACPs, genetic or epigenetic mechanisms have been documented, resulting in either Wnt pathway activation or in other potential targetable pathways downstream of the Wntβ-catenin pathway.
The involvement of the MAPK/ERK pathway in CP pathogenesis implicates MEK inhibitors as candidate drug mediators.30 Patel et al. described the beneficial effect of off-label binimetinib in a multi-treated 26-year-old female with ACP along with hypothalamic obesity, insulin-independent diabetes mellitus, hepatic cirrhosis with secondary splenomegaly and cytopenias, and stasis dermatitis.61 She started at 45 mg twice daily with occasional brief drug interruptions, with dose reductions to 30 mg twice daily and subsequently to 30 mg in the morning and 15 mg in the evening. Brain magnetic resonance imaging (MRI) 8 months after treatment showed a remarkable decrease in tumour size (from 3.5×2.5×2.5 cm to 2.8×2.1×1.8 cm), which remained stable at least for another 8 months. A number of AEs where reported, such as furuncles/papulopustular rash on thighs and buttocks, nail dystrophy, hyponatraemia, venous stasis with poor wound healing, fatigue/daytime sleepiness and weight gain. However, it is not clear whether these side effects were associated with the drug or with previous therapies, since the patient was overtreated with eight surgical resections, chemotherapy (vinorelbine), radiation therapy and γ-knife radiosurgery (Table 1, patient 23).
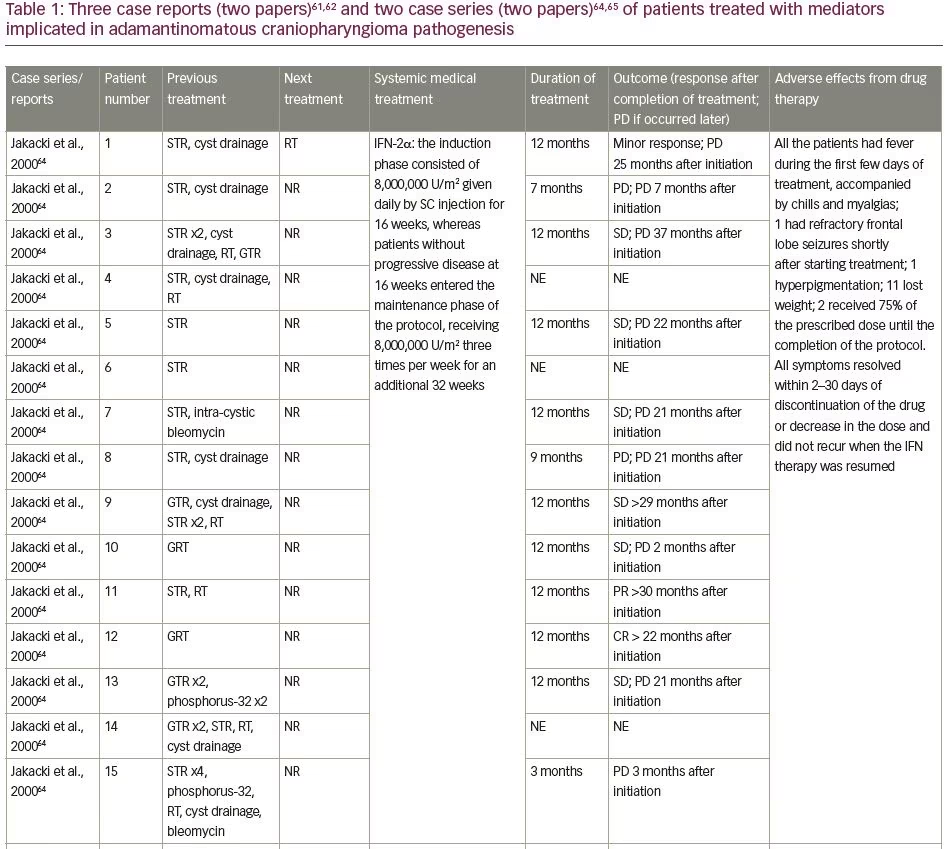
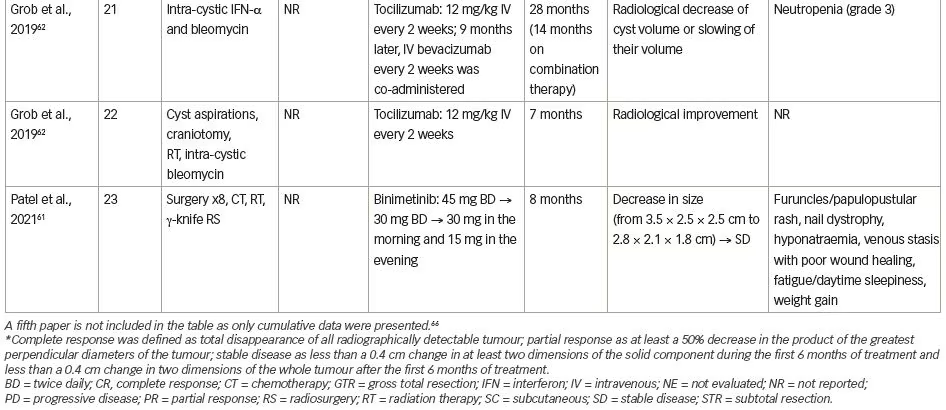
Immune system-targeted therapy is another promising therapeutic target for ACPs, since the paracrine action of inflammatory mediators has been identified both in the solid and cystic tumour compartments. Tocilizumab, a humanized monoclonal antibody against soluble and membrane-bound IL-6R,62 was offered to a 4-year-old boy with ACP. He had 14 months of therapy with intra-cystic interferon-α followed by intra-cystic bleomycin via a cyst catheter; however, both failed to control his disease. Tocilizumab 12 mg/kg was given intravenously (IV) every 2 weeks on a compassionate basis for 6 months, and was well tolerated. A radiological improvement (reduction in size, or slowed growth of cyst) was seen 6 months following treatment. In the ninth month of treatment, the combination therapy of IV tocilizumab and IV bevacizumab every 2 weeks was initiated and was generally well tolerated. In total, he received 14 months of combination therapy over 28 months, with a significant radiological reduction in cysts’ size, followed by tumour surveillance because of stable disease. Few AEs delayed the treatment, which were mostly represented by neutropenia (Common Terminology Criteria for Adverse Events [CTCAE] v5, Grade 3) (Table 1, patient 21). Tocilizumab was also given to a 7-year-old boy as compassionate monotherapy after multiple cyst aspirations followed by craniotomy, radiotherapy and intra-cystic bleomycin, which resulted in a radiological improvement. Even in this latter case, the drug was given for 7 months, followed by image monitoring (Table 1, patient 22).62 The efficacy of tocilizumab in the management of ACP is being studied in a phase 0, open-label clinical study investigating its administration every 2 weeks for up to 2 years (ClinicalTrials.gov Identifier: NCT03970226).
The primarily studied immunotherapy evaluated in ACP is interferon. Interferon is a small molecule that modulates host immune response, also displaying direct anti-proliferative and cytotoxic effects in tumoural cells.63 The first study to systematically administer interferon studied 15 children aged ≤21 years who received subcutaneous, short-acting interferon daily for 16 weeks, which was followed by a 32-week maintenance phase, where interferon was given three times per week.64 Three out of 12 (25%) patients who were evaluated had a documentable response (complete, partial or minor), whereas one patient showed a clinical response (visual fields improvement). The median time to progression was 25 months.
A 6-year-old girl, having previously undergone STR and cyst drainage, showed an increase in cyst size during the first 4 months of therapy, followed by a cyst size reduction without any additional intervention. When the treatment was complete, the neoplasm had reduced by 45%, but 13 months post-treatment, the patient developed a radiological progression and received external-beam radiotherapy (Table 1, patient 1). In a 13-year-old boy, interferon treatment was offered after STR and radiotherapy (5,130 cGy), with progressive radiological shrinkage of the tumour and significant visual improvement. When the treatment was completed, the neoplasm had been reduced by 75%, and the patient had clinically and radiologically stable disease 18 months post-therapy (Table 1, patient 11). A 13-year-old boy received interferon after GTR; a progressive radiological decrease in tumour size was documented. Nine months post-therapy, no residual tumour could be seen, which was retained for more than 10 additional months post-therapy (Table 1, patient 12). Overall, 6 patients (50%) showed increases in the cystic component with the initial interferon administration, with 2 patients having further increases in the cystic component, displaying progressive disease, and another 2 patients having stable disease with no further increase of cyst size.64
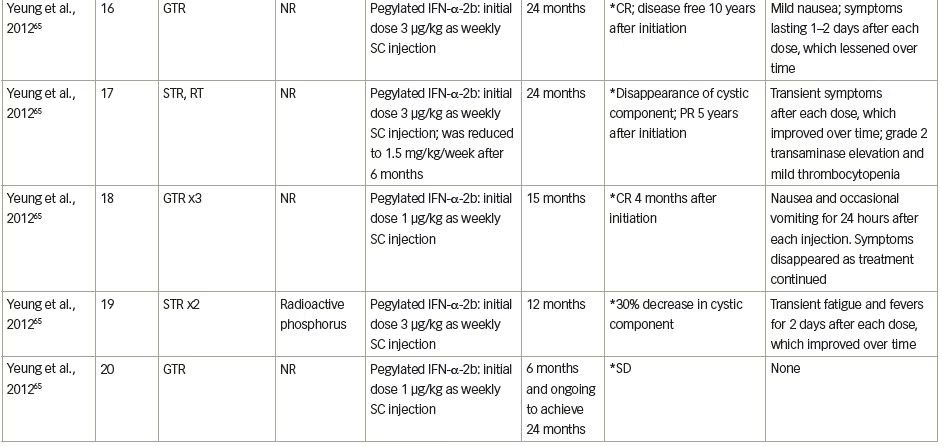
The next step was to investigate a better way to deliver the interferon. Pegylated interferon has a longer half-life and more sustained action than non-pegylated interferon, and was used in a recent pilot study in five children with ACP.65 In this study, the first case, a 9-year-old girl, underwent GTR, but had cyst recurrence 3 months later. Under the interferon therapy, the cyst initially increased in size, but then decreased, followed by a complete disappearance at the end of the therapy. No additional therapy was needed, and the patient remained free of disease 8 years post-interferon therapy (Table 1, patient 16). The second case, a 14-year-old boy, had previously had STR, followed by radiotherapy (5,400 cGy); when the residual partially cystic tumour increased in size, interferon was started. The cystic component of the ACP completely disappeared 4 months post-treatment, whereas the residual calcified mass remained stable 5 years later (Table 1, patient 17).
A 13-year-old boy previously treated with three GTRs had cyst recurrence and interferon was started. Seven months later, he had a complete radiological response that he maintained during the whole 15-month treatment period (Table 1, patient 18). A 14-year-old boy previously treated by a two-staged STR started interferon treatment when the cystic component of the neoplasm increased. Four months post-treatment, the cystic component had reduced by 30%, whereas 12 months post-therapy initiation, of the solid and cystic components, only the cystic component of the neoplasm increased; the patient was referred for treatment with radioactive phosphorus (Table 1, patient 19). Finally, a 15-year-old girl had been previously treated with GTR, and after recurrence, interferon treatment was initiated. She had stable disease 6 months post-treatment, with intention to be treated until 24 months (Table 1, patient 20). Overall, a complete response was seen in 40% of the children treated, 40% had a partial response and 10% had a stable disease, with no major safety concerns. Despite the fact that 80% of patients had AEs, these were transient and disappeared as treatment continued.
Based on this study, a phase II study of peginterferon alfa-2b for patients with unresectable or recurrent CPs was performed; 7 patients underwent surgery alone and 11 previously received radiotherapy, aged 18 months–25 years.66 Against expectations created by the previous trials, the objective responses were limited but the progression-free survival was protracted. Two of the 7 patients (28.6%) who had surgical treatment had a partial response, but only 1 had a radiological improvement for more than 3 months. Five out of 11 patients previously treated with radiotherapy received at least 1 year of interferon, but none showed an objective radiographic response.
Following these observations extracted from the five aforementioned publications, a number of additional clinical trials are currently running. These ongoing clinical trials are investigating the effectors of Wnt pathway inhibition (such as XNW7201, CGX1321, RXC004) in adult patients with solid tumours, as monotherapy or combination therapy (ClinicalTrials.gov identifiers: NCT03901950, NCT02675946, NCT03447470). Hence, the results from the phase I study to evaluate the safety, tolerability and pharmacokinetic profile of XNW7201, a new medication that blocks the activity of the Wnt protein in subjects with advanced solid tumours, will unravel a new field in pharmacological management of ACPs (ClinicalTrials.gov identifier: NCT03901950). Nevertheless, concerns about eventual serious AEs with targeting these pathways have been raised.17,35,67 IDO-1, an immunosuppressant metabolic enzyme, represents an eventual druggable target,68 and the IDO-1 inhibitors epacadostat (INCB024360) and navoximod (GDC-0919) are being investigated in clinical trials.69,70
Systemic medical therapy in papillary craniopharyngiomas
The presence of p.BRAF-V600E mutations in patients with PCPs has provided a molecular rationale for the use of MAPK/ERK pathway inhibitors in these patients by using BRAF-V600E with or without MEK inhibitors.17 Firstly, vemurafenib (960 mg twice daily) was given off-label to a 53-year-old female with a 26-year history of a recurrent PCP, with a proven BRAF-V600E mutation and progressive visual failure following three trans-sphenoidal operations and fractionated radiotherapy.71 An early radiological reduction in tumour size, along with clinical improvement, was seen 2 weeks post-treatment; near complete remission was documented 3 months post-treatment. However, the brisk reduction in tumour size resulted in cerebrospinal fluid leaking, with pneumocephalus and meningitis which were treated with antimicrobial therapy and surgical repair. The drug was discontinued, but the tumour recurred 6 weeks later and vemurafenib was re-started; there was a re-reduction of tumour size followed by stabilization for 7 months. Unfortunately, a regrowth was documented in this case, and treated with vemurafenib (Table 2, patient 1).
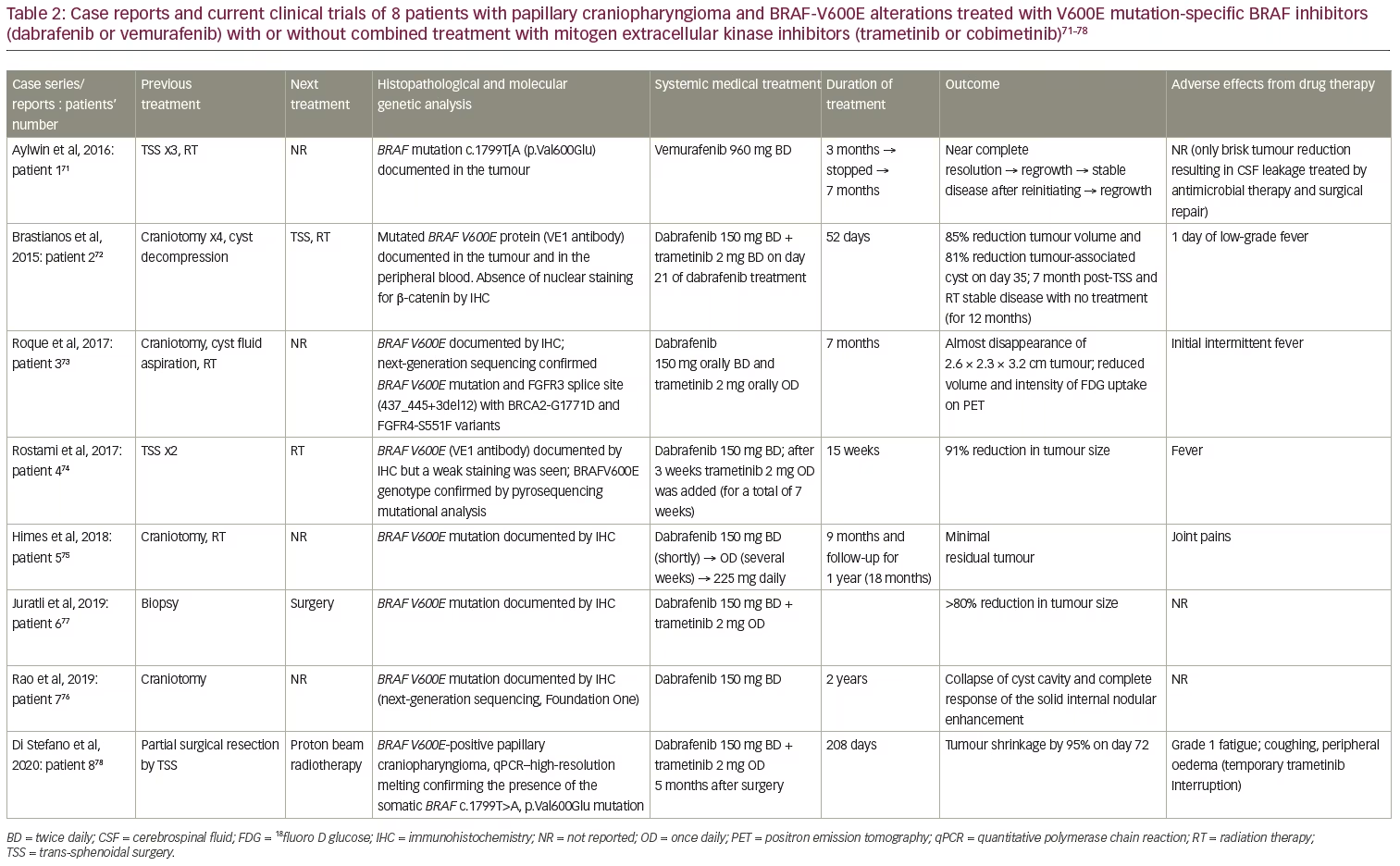
In parallel, dabrafenib (150 mg twice daily) was given to a 39-year-old male with a PCP and documented BRAF-V600E mutation (Table 2, patient 2).72 This patient had undergone four craniotomies along with cyst decompressions, though the aggressive clinical behaviour of the tumour stressed the need for additional treatment. A 23% reduction in tumour size and a 32% decrease in cyst volume were identified as early as 4 days post-treatment, followed by a 52% reduction in tumour size and 70% decrease in cyst volume 17 days after treatment. Trametinib (2 mg, orally, twice daily) was added on day 21. An 85% reduction in tumour size and an 81% decrease in cyst volume was documented 14 days after the combined treatment. The patient underwent endoscopic trans-sphenoidal surgery on day 38 of treatment, and on day 52, the medical treatment was discontinued, followed by radiotherapy 1 week later (5,040 cGy in 28 fractions). This patient remained free of new symptoms at the 7-month follow-up.72
A 47-year-old female with documented PCP and BRAF-V600E mutation presented with a right frontal craniotomy to remove a 2.7 cm cystic lesion, but the resection was limited due to tight adherence of the tumour to the optic apparatus. A residual tumour of 1.1 cm did not give her new symptoms.73 Ommaya catheter placement and radiotherapy (5,400 cGy in 30 fractions) followed because of tumour recurrence and worsening vision. After further clinical and radiological deterioration 7 months post-diagnosis, dabrafenib (150 mg twice daily) and trametinib (2 mg once daily) were administered. Two months later, a response was documented and 7 months post-treatment, the tumour was nearly radiologically disappeared, alongside a clinical improvement (Table 2, patient 3).73 Moreover, a 65-year-old male with visual deficits had two subsequent STRs of a PCP with documented BRAF-V600E mutation.74 Rapid tumour growth before radiotherapy prompted administration of dabrafenib (150 mg twice daily), and 3 weeks later, trametinib (2 mg once daily for 7 weeks). An 11% tumour reduction was seen early at 4 weeks, and a 91% reduction at 15 weeks of treatment while waiting for radiotherapy (Table 2, patient 4).
These four cases showed that monotherapy was then changed to combination therapy with even better results. However, a fifth case showed the efficacy of long-term treatment with dabrafenib monotherapy in a 47-year-old male patient with a previous medical history of non-Hodgkin’s lymphoma and colon cancer, who had undergone craniotomy and radiotherapy (3,600 cGy in 12 fractions) for a PCP with confirmed BRAF-V600E mutation. The patient received three chemotherapeutic schemes and local radiotherapy targeting the hypermetabolic left supraclavicular lymph nodes for his colon cancer. However, 3 years post-radiotherapy, his PCP recurred and dabrafenib was initiated (150 mg twice daily, reduced to 150 mg once daily due to joint pains, and then increased to 225 mg daily). After 2 months of treatment, the cystic component enlarged, while the solid component reduced in size. Six months post-treatment, a significant reduction in size of both components was seen, and by 9 months, only a minimal residual tumour was evident, with stable disease 12 months post-treatment. Treatment was discontinued, and the tumour remained stable for a follow-up period of 12 months (Table 2, patient 5).75
Similarly, the longest trial of dabrafenib monotherapy has been recently published, achieving a 2-year treatment period.76 A 35-year-old male patient underwent craniotomy with residual BRAF-V600E mutant PCP tumour, and a course of combined treatment with dabrafenib and trametinib was requested because of a rapid deterioration. However, only dabrafenib was approved as an off-label treatment. Two months after starting treatment, the patient had clinical and radiological improvement, with decreased size, enhancement and oedema of the residual suprasellar CP. Two years later, MRI showed the continued collapse of the patient’s cyst cavity and complete response of the solid internal nodular enhancement, along with a continued clinical improvement (Table 2, patient 7).
The neoadjuvant role of this druggable approach has been also investigated. A BRAF-V600E mutant PCP was diagnosed in a 21-year-old male during the first surgery of a mass proved to be a PCP, which was interrupted since it was complicated by arterial infarcts. Dabrafenib and trametinib treatment for a 6-month period significantly reduced tumour size by 80% and 90% in the solid and cystic portions, respectively (Table 2, patient 6).77 In addition, the combination of dabrafenib and trametinib was also given as neoadjuvant treatment to proton beam radiotherapy (PBRT). A 55-year-old woman had only a 15% tumour debulking due to intra-operative bleeding and had a rapid deterioration 3 months later. Starting dabrafenib 150 mg twice daily combined with trametinib 2 mg once daily 5 months after surgery reduced the tumour size by as much as 94.5% after 72 days of treatment, followed by tumour size stabilization. The overall duration of medical treatment was 208 days, followed by 7 days of no treatment with an increase of the cystic component of the tumour (13% increase, corresponding to 0.7 cm3), which further progressed to 1.5 cm3 (and total volume of 1.5 cm3) followed by stabilization. A total follow-up at 18 weeks after the end of PBRT (5,220 cGy in 29 fractions) showed a volume reduction of 0.14 cm3 and total volume of 1.5 cm3 (Table 2, patient 8).78
All these excellent results in the patient with PCP led to a clinical trial in patients with BRAF-V600E-positive PCP, using a combination of vemurafenib (BRAF-V600E inhibitor) and cobimetinib (a MEK inhibitor) (ClinicalTrials.gov Identifier: NCT03224767). In this ongoing open-label, phase II cooperative group trial (Alliance A071601), vemurafenib is administered twice daily on days 1–28, and cobimetinib four-times daily on days 1–21, as recently described;79 treatment is repeated every 28 days for up to five cycles in the absence of disease progression or unacceptable toxicity, and thereafter patients may be treated with radiotherapy, surgery or continuing the combination of medical treatment.
Conclusions
A benign disease such as CP needs management that is effective and at the same time safe, since patients may lead long lives, and treatment complications may alter their quality of life, morbidity and mortality. Molecular pathophysiology advances are the main tool in achieving this target. The potential benefits of BRAF inhibitors, achieving either transient tumour response, enabling their use as neoadjuvant treatment or following recurrence, have opened the horizons for intensive investigation in the field of managing PCPs. More recently, the use of MEK inhibitors and immune system therapy in ACP is a promising medical therapy for this large subset of CP patients. A number of ongoing trials will shed light on schemes, doses, combined treatments and safety issues of new molecular-targeted treatments, and will change the management of CPs by launching the era of personalized medicine in these rare neoplasms.


