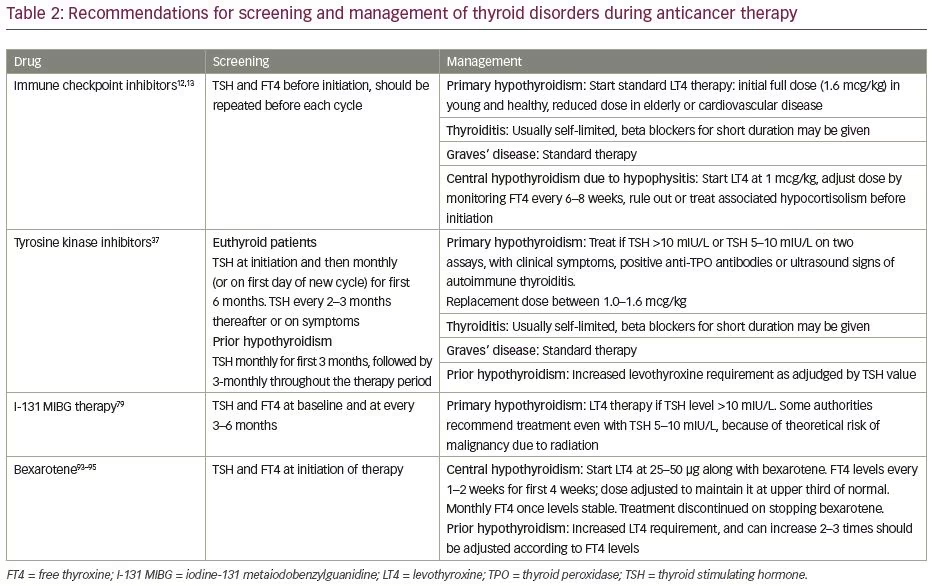
The armamentarium of anticancer drugs available to an oncologist has grown rapidly over the past few decades. The use of cancer immunotherapy and targeted therapy has become more popular in the last few years. It has also become increasingly clear that various anticancer agents, both conventional and newer ones, may be associated with certain off-target adverse effects involving the endocrine system, especially the thyroid gland.1 The site of action of commonly used cancer immunotherapy agents is depicted in Figure 1. This article is aimed at describing thyroid dysfunction associated with various anticancer agents. These have been briefly summarised in Table 1. It is important to identify and appropriately treat thyroid dysfunction in such patients. This will not only improve their overall quality of life, but also ensure optimal treatment outcome.
Literature search
A MEDLINE search was conducted for articles published before 30 April 2019. Articles published in English were considered. The search terms were words related to thyroid disorders, such as ‘thyroid’, ‘hypothyroidism’, ‘thyrotoxicosis’, ‘hyperthyroidism’ ‘Graves’ disease’ ‘central hypothyroidism’, and ‘thyroiditis’ in association with ‘anticancer drugs’, ‘immune checkpoint inhibitors’, ‘tyrosine kinase inhibitors’, ‘interferon-α,’ ‘interleukin-2,’ ‘alemtuzumab’, ‘thalidomide’, ‘lenalidomide’, ‘pomalidomide’ ‘radioiodine-based cancer therapies’ and ‘bexarotene’. The names of specific drugs, like ipilimumab, nivolumab, pembrolizumab amongst immune check point inhibitors; and sunitinib, sorafenib, axitinib, pazopanib, vandetanib, motesanib, imatinib, cabozantinib, nilotinib, dasatinib, erlotinib, gefitinib, lapatinib, nintedanib, regorafenib and tivozanib amongst tyrosine kinase inhibitors, were also included in the search. The abstracts were evaluated for relevance, and full text of all appropriate articles was retrieved. Reference lists of selected articles were also searched. Articles describing usage of anticancer agents for treatment of thyroid cancer were excluded.
Immune checkpoint inhibitors
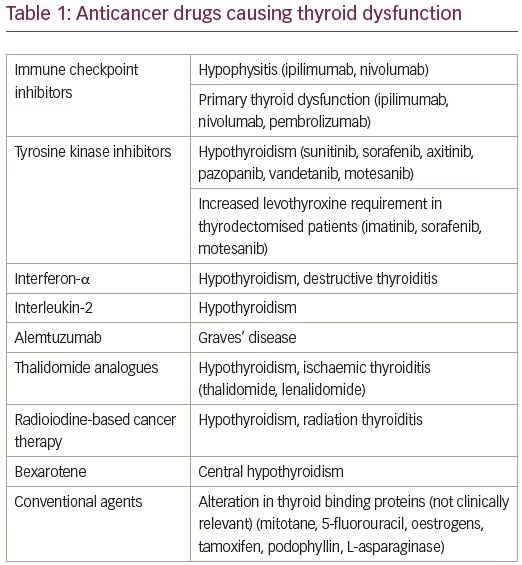
A better understanding of the complexities of the human immune system and its regulation paved the way for a novel concept in the field of oncology termed ‘cancer immunotherapy’. The basic principle of cancer immunotherapy is utilisation of body’s own immune system to target cancer cells. Immune checkpoint molecules are regulators of the immune system which provide self-tolerance and prevent the immune system from destroying its own cells (Figure 2). These include cytotoxic T-lymphocyte associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1; a cell surface receptor) and its ligand (PD-L1). CTLA-4 is constitutionally expressed on regulatory T cells, gets up-regulated on T-cell activation, and works toward inhibiting a second (co-stimulatory) signal for T-cell activation. PD-1 is present on T cells, B cells and natural killer (NK) cells, and binds to PD-L1 expressed by tumour cells. The interaction between PD-1 and PD-L1 inhibits destruction of tumour cells by the immune system; hence, PD-L1 is over expressed by the tumour cells to their advantage.2,3
Indications and adverse events
Immune checkpoint inhibitors are agents that inhibit these immune checkpoint molecules, causing an immune attack on the tumour cells. The Food and Drug Administration (FDA)-approved immune checkpoint inhibitors include anti-CTLA-4 monoclonal antibody (ipilimumab), anti-PD-1 monoclonal antibody (nivolumab, pembrolizumab), and anti-PD-L1 monoclonal antibody (atezolizumab, avelumab, and durvalumab). While initially approved for the treatment of unresectable or metastatic melanoma, these agents have been found to be effective in a wide variety of tumours, such as small-cell lung cancer, non-small-cell lung cancer (NSCLC), Hodgkin’s lymphoma, renal cell cancer (RCC), prostate cancer, bladder cancer, oesophageal cancer and breast cancer.4 The enhanced immune activation seen with these agents can lead to a variety of immune-related adverse events. These may involve a number of organ systems – skin (rash, pruritus), gastrointestinal tract and liver (colitis, autoimmune hepatitis), and the endocrine system (hypophysitis and thyroid dysfunction). In terms of endocrine dysfunction, hypophysitis has been reported more often with CTLA-4 inhibitors, while thyroid dysfunction has been reported with both CTLA-4 and PD-1/PD-L1 inhibitors.4
Hypophysitis and central hypothyroidism
Hypophysitis related to CTLA-4 inhibitor therapy has been reported at an incidence of 0.4–17.0% in literature.5–7 Similar to autoimmune lymphocytic hypophysitis, there is predilection for involvement of thyrotropes (central hypothyroidism) and corticotrophs (central hypoadrenalism). However, unlike lymphocytic hypophysitis, CTLA-4 inhibitor-related hypophysitis is more common in males. A possible reason could be the higher proportion of male participants in studies reporting hypophysitis with CTLA-4-inhibitor therapy. In a retrospective study involving 154 adult subjects with melanoma treated with ipilimumab at a tertiary care centre in USA, hypophysitis was reported at a prevalence of 11% (17/154).6 The prevalence was significantly higher in males compared to females (15.6% versus 3.6%) and subjects with hypophysitis were significantly older compared to the remaining cohort (mean age 68.2 versus 59.9 years). The diagnosis of hypophysitis was made after a median duration of 8.4 weeks following the initiation of ipilimumab. Central hypothyroidism was reported in all, while central hypoadrenalism was seen in 42% (7/17) subjects with hypophysitis. Interestingly, the median survival in subjects who developed hypophysitis was significantly higher than those who did not (19.4 versus 8.8 months). In another study by Ryder et al. (n=254), the overall incidence of hypophysitis following ipilimumab therapy was 8%, while in the subjects receiving combination immune checkpoint-inhibitor therapy (ipilimumab and nivolumab), the incidence increased to 9%.7
Primary thyroid dysfunction
Primary thyroid dysfunction following immune checkpoint-inhibitor therapy has been described in the form of subclinical or overt hypothyroidism, transient thyrotoxicosis and painless thyroiditis.4–7 Rare cases of Graves’ disease and euthyroid orbitopathy have also been described.8–10 The incidence of thyroid dysfunction following immune checkpoint-inhibitor therapy has been variably reported at 1–22% in the literature.4,7,8–11 The incidence is much higher in patients receiving a combination therapy (CTLA-4 inhibitor and PD-1 inhibitor). In the study by Ryder et al., the overall incidence of hypothyroidism/thyroiditis following ipilimumab was 6%, while it increased to 22% in those receiving a combination of ipilimumab and nivolumab.7
In the KEYNOTE-001 study, 51 subjects with advanced NSCLC treated with PD-1 inhibitor pembrolizumab were studied for thyroid dysfunction. Of the 48 subjects with euthyroidism at baseline, 10 (21%) developed hypothyroidism (requiring replacement) at a median duration of 6 weeks. Hypothyroidism was preceded by a phase of transient thyrotoxicosis in 6/10 (60%) subjects. Anti-thyroid antibodies were more frequent in the subset of subjects who developed thyroid dysfunction (8/10, 80%), compared to those who did not (3/38, 8%).11 Similar to the ipilimumab hypophysitis study, subjects developing thyroid dysfunction had overall better survival (hazard ratio, 0.29; 95% confidence interval 0.09–0.94; p=0.04). The authors concluded that primary thyroid dysfunction following pembrolizumab therapy is fairly common, occurs early, is associated with transient thyrotoxicosis and anti-thyroid antibodies in most cases, and possibly portends a favourable outcome.
The association between immune checkpoint inhibitor-related endocrine dysfunction and better overall survival seen in various studies might indicate higher immune activation and better tumour-cell destruction by the immune system in subjects developing this adverse effect. This interesting observation needs further validation with well-designed, long-term follow-up studies.
Management
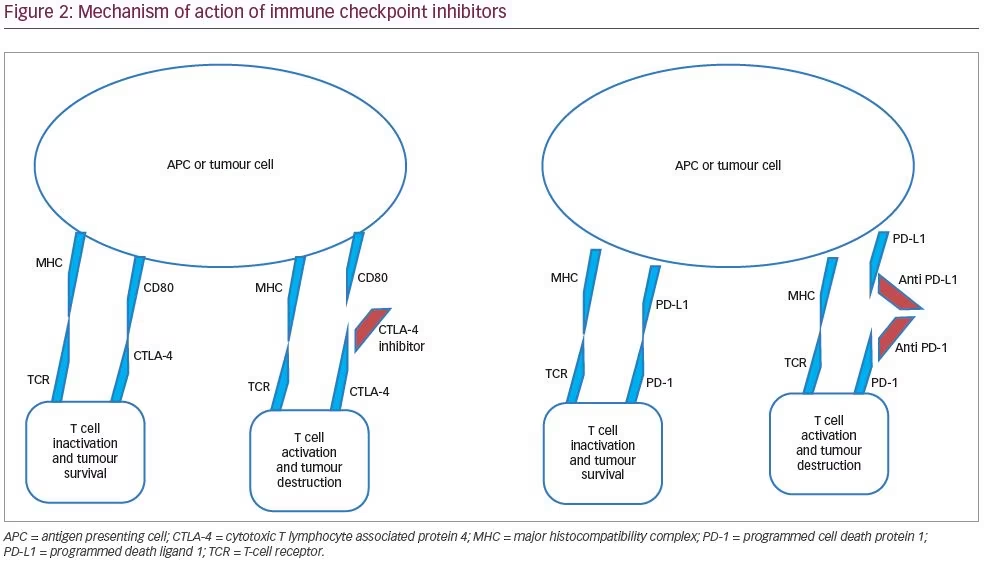
The mainstay of management of thyroid dysfunction following immune checkpoint-inhibitor therapy involves appropriate thyroid hormone replacement.12,13 In patients with combined adrenal and thyroid dysfunction due to immune checkpoint inhibitor-related hypophysitis, thyroid hormone replacement should begin 3–5 days after initiation of glucocorticoid replacement in order to avoid an acute adrenal crisis. Endocrine dysfunction following immune checkpoint inhibitor-related hypophysitis is generally irreversible, and high-dose glucocorticoid should only be considered in patients with significant mass effects or severe hyponatremia.14,15 In terms of primary thyroid dysfunction, subclinical hypothyroidism (with normal free thyroxine [FT4] and thyroid stimulating hormone [TSH] <10 mIU/L) could be managed with careful observation and close follow-up.4 Thyrotoxicosis is typically transient (ultimately giving way to primary hypothyroidism), and may be treated with beta blockers for a brief duration. Radioiodine scan may help to differentiate thyrotoxicosis related to thyroiditis from hyperthyroidism related to Graves’ disease. However, one should be cognizant of the fact that such patients frequently undergo iodinated contrast-based imaging for their primary disease. This may interfere with the uptake of radioiodine by saturating the thyroid gland iodine pool.
Tyrosine kinase inhibitors
Tyrosine kinase inhibitors (TKIs) are one of the most potent classes of targeted therapy drugs, used in many different cancer types. They inhibit the enzyme tyrosine kinase, which is involved in transfer of phosphate from adenosine triphosphate to tyrosine residues in the catalytic domain of growth factor receptors. TKIs by this mechanism inhibit vascular endothelial growth factor receptors (VEGFRs) and their downstream targets and suppress endothelial proliferation. Prevention of vascular growth in the tumour causes disruption of the supply of nutrients and oxygen and kill the tumour cells.16
Indications and thyroid dysfunction associated with tyrosine kinase inhibitors
Currently, the FDA has approved more than 20 different TKIs for clinical use.17 In comparison to traditional cytotoxic antineoplastic agents, TKIs have high selectivity, high efficacy, low side effects and have superiority in the treatment of chronic myeloid leukaemia, NSCLC and RCC. They are also active against medullary thyroid cancer and differentiated thyroid cancer refractory to iodine-131 (I-131), gastrointestinal stromal tumour (GIST), pancreatic neuroendocrine tumours and hepatocellular carcinoma.16 Thyroid dysfunction is recognised as an important but manageable side effect of TKIs. Although all TKIs act through the same mechanism, they differ in their spectrum of targeted kinases, thereby resulting in varying rate of thyroid dysfunction. TKIs can induce de novo hypothyroidism which can be preceded by a phase of transient thyrotoxicosis in 20–40% cases.18 The TKIs which are known to induce new onset hypothyroidism in a significant proportion of patients include sunitinib, sorafenib, axitinib, pazopanib, vandetanib and motesanib.19 The other variety of thyroid dysfunction induced by TKI is increased levothyroxine (LT4) requirements in patients who have been on stable doses of thyroid hormone replacement following thyroidectomy for other reasons.20
Sunitinib induced thyroid dysfunction
The TKI most frequently associated with development of new onset hypothyroidism is sunitinib.21–26 It is approved for the treatment of RCC, and GISTs resistant to therapy with imatinib. Hypothyroidism occurs in 14–70% of recipients in different studies.21–23 A meta-analysis of seven randomised trials which included 2,787 subjects revealed a risk ratio for all and high-grade hypothyroidism of 13.95 and 4.78, respectively.24 The risk of developing hypothyroidism increases with time and with the number of cycles of therapy.25 The time for development of thyroid dysfunction in the two largest series with long follow-up was found to vary from as early as 4 weeks to as late as 92 weeks.25,26
Thyroid dysfunction induced by other tyrosine kinase inhibitors
Sorafenib is used for the treatment of patients with metastatic RCC, advanced hepatocellular carcinoma and radioactive iodine-resistant advanced thyroid cancer. The incidence of thyroid dysfunction with sorafenib is much less when compared to sunitinib, ranging between 6.3–27.0%.27–29 The median time to develop hypothyroidism was 20 months, but an increase in TSH could appear as early as 6 weeks after initiation of treatment.27,29 Axitinib, used for treatment of RCC, in the phase III AXIS trial, caused hypothyroidism more commonly compared to sorafenib (21% versus 7%).30 Pazopanib, a TKI approved for the treatment of RCC and soft tissue sarcoma, has reported rates of hypothyroidism of around 12% or less.31 Other TKIs linked with hypothyroidism include cabozantinib, nilotinib, dasatinib, erlotinib, gefitinib, lapatinib, nintedanib, regorafenib and tivozanib.17
Mechanism of tyrosine kinase inhibitor-induced hypothyroidism
The pathophysiology behind development of hypothyroidism is presumed to be vascular, resulting from its anti-angiogenic effect. The thyroid gland is a highly vascular organ and its blood flow is mainly dependent on the VEGFR signalling. Broad spectrum TKIs like sunitinib which not only inhibits VEGFR2 and VEGFR3, but also VEGFR1 and the platelet-derived growth factor receptor (PDGFR) impairs vascularisation of thyroid and induces thyroid ischemia.32 However, PDGFR signalling does not play a role in thyroid angiogenesis in physiological condition, but becomes active in setting of ischaemia. The compensatory mechanism to restore vascularity might get impaired when it is inhibited.33 The differential effect of various TKIs in inducing thyroid dysfunction can be attributed to their selectivity to block diverse vascular growth factor signalling pathways. A rapid reduction in thyroid vascular flow can result in ischaemic thyroiditis and produce the preceding transient thyrotoxic phase.34 Interestingly, hypothyroidism resulting from TKIs has been associated with longer survival for unclear reasons.35
Tyrosine kinase inhibitors in thyroidectomised patients
Another type of thyroid dysfunction occurs in thyroidectomised patients who have worsening of stable hypothyroidism and increased LT4 requirement after initiating TKI therapy. Imatinib, currently used in treatment of chronic myeloid leukaemia, GISTs and other haematological cancers, is most commonly associated with this phenomenon. Imatinib does not induce hypothyroidism in those with intact thyroid gland.20 Sorafenib and motesanib have also been reported to cause similar elevation in TSH in patients with pre-existing hypothyroidism.36,37 The possible mechanisms could be stimulation of type 3 deiodinase activity leading to increased peripheral inactivation of thyroid hormones and a dose-dependent inhibition of thyroid hormone transport protein, monocarboxylate transporter.35,38 Additionally, reduced pituitary type 2 deiodinase activity can cause intracellular depletion of triiodothyronine (T3) in the thyrotrophs and an inappropriate elevation of TSH for the concomitant serum free T3 and FT4 levels.32
Screening and management of tyrosine kinase inhibitor-induced thyroid dysfunction
Drui et al. recommends screening by measurement of TSH at initiation of TKI treatment and then on a monthly basis (or on first day of a new cycle in cases of interrupted treatment) for the first 6 months. Thereafter, TSH estimation should be done every 2–3 months or in cases of clinical signs of thyroid disorder. In individuals with pre-existing hypothyroidism, TSH should be monitored monthly for first 3 months, followed by 3-monthly intervals throughout the therapy period. LT4 should be initiated if TSH is >10 mIU/L or if TSH is between 5–10 mIU/L on two assays along with clinical symptoms, presence of anti-thyroid antibodies or ultrasound evidence of autoimmune thyroiditis. After the end of TKI treatment, a trial of withdrawal of LT4 should be considered with appropriate monitoring.39
Other Immunomodulators
Interferon-α
Interferon-α (IFN-α) is a cytokine approved for treatment of hepatitis C virus (HCV) and several kinds of malignancies like melanoma, RCC, Kaposi’s sarcoma. It is also used for treatment of hairy cell leukaemia and follicular lymphoma. In recent years it has largely been replaced by better alternatives as an anticancer agent. It exerts direct antitumor effect and also indirectly causes immune mediated destruction of tumour cells by inducing expression of major histocompatibility complex 1 (MHC-1), tumour specific antigen and adhesion molecules on the cell surface.40
The mechanism of IFN-α-induced thyroid dysfunction (IITD) is not fully understood. The expression of MHC-1 antigens on cell surface result in activation of cytotoxic T cells which in turn cause cellular destruction.41 The presence of pre-existing intrathyroidal lymphocytes thereby increases the susceptibility for the development of IITD. This also explains the finding of higher likelihood of development of thyroid dysfunction in individuals with anti-thyroid antibodies.42,43 Mandac et al. classified IITD in patients with HCV into two groups: autoimmune and nonautoimmune.44 Obołon´czyk et al. suggested the addition of a new term, ‘undifferentiated IITD’, in view of variable course of the disease.45 The full spectrum of IITD described in patients with HCV may be seen in those receiving IFN-α therapy for malignant conditions.46
Clinical thyroid disease is found in 15–20% of patients with HCV receiving IFN-α, and up to 40% of those become thyroid-antibody positive.45,47 Autoimmune hypothyroidism is the most common clinical presentation, occurring in 20% of cases followed by destructive thyroiditis (2–3%). Graves’ disease is found rarely.48 Notably, HCV infection itself can induce thyroid dysfunction. IFN-α treatment for malignancy carries a lesser chance of development of thyroid dysfunction as compared to patients with HCV.44 Thyroid disorders occur in 2.4–31.0% of patients receiving IFN-α therapy for solid tumors.46 It is recommended to screen for thyroid dysfunction with TSH and antithyroid antibodies prior to starting IFN-α. Follow-up TSH should be done every 2 months in those with positive antithyroid antibody and every 6 months in those with negative antibody status.1
Interleukin-2 (aldesleukin)
Interleukin-2 (IL-2) is another cytokine-based therapy used for treatment of metastatic melanoma and RCC. Its use has declined in recent years after availability of better tolerated alternatives. IL-2 destroys tumour cells by activating NK cells and antigen-specific T cells.49
In a large cohort of 281 patients, treated with IL-2 alone, hypothyroidism was reported in 35% of patients, although only 9% of patients required LT4. Longer duration of therapy correlated with higher risk of development of hypothyroidism. Thyrotoxicosis occurred in 7% of patients receiving high-dose IL-2. Overall incidence of thyroid dysfunction did not differ in the high- and low-dose IL-2 arms.50 In data from PROCLAIMSM, a registry of patients receiving high-dose IL-2, 70% of patients developed primarily vitiligo (in cases of melanoma only) and/or thyroid dysfunction (greater incidence in patients with RCC compared to patients with melanoma). The development of these immune-related adverse events correlated with improved tumour control and overall survival.51 Hypothyroidism usually ensues by 4–17 weeks after starting IL-2 and may be reversible on discontinuation of treatment.52,53 Frequency of thyroid dysfunction is 10–60% in various reports but in many of these studies patients received IL-2 in combination with IFN-α, lymphokine-activated killer cells, or vaccine.46
IL-2 stimulates autoreactive lymphocytes and induces thyroid autoimmunity. It also increases IL-1, tumour necrosis factor alpha (TNF-α), and IFN-γ. These cytokines induce the presentation of human leukocyte antigen class II (HLA-II) and associated autoantigens on thyrocytes and causes its autoimmune destruction.54,55 High levels of thyroid antibodies prior to treatment amplified the risk of IL-2 induced hypothyroidism.53 TSH should be measured before initiating treatment with IL-2 and then once every 2–3 months during therapy.1
Alemtuzumab
Alemtuzumab is a recombinant humanised monoclonal antibody that is directed against the cell surface glycoprotein, CD52 present on lymphocytes. It causes profound lymphopenia by causing complement mediated lysis of these cells. The change in immune repertoire that occurs during subsequent lymphocyte reconstitution accounts for its therapeutic effect.56 It is administered for the treatment of T cell prolymphocytic leukaemia and chronic lymphocytic leukaemia (CLL). Alemtuzumab is also approved for treatment of active relapsing-remitting multiple sclerosis (MS). It is used as an immunosuppressive agent following solid organ and stem cell transplant and in graft versus host disease.
Thyroid dysfunction commonly occurs after therapy with alemtuzumab for MS, but has not been reported after treatment of malignant conditions.57–59 In a recently published study, thyroid dysfunction occurred in 41% of 248 MS patients treated with alemtuzumab. Graves’ disease accounted for 72% of them with a median onset of 17 months following the last dose, and the majority (89%) within 3 years.57 In another large series of 334 patients with MS, thyroid dysfunction was reported in 34% of patients receiving alemtuzumab as compared to 6.5% receiving IFNβ-1a. Graves’ disease was reported in 22%, hypothyroidism in 7%, and subacute thyroiditis in 4%. Thyroid binding inhibitory immunoglobulin was positive in 74% of patients with overt hypothyroidism.58 Patients receiving alemtuzumab for T cell prolymphocytic leukaemia and chronic lymphocytic leukaemia did not develop thyroid dysfunction.59,60 Autoimmunity following alemtuzumab therapy has been attributed to a breakdown in self-tolerance during immune reconstitution and is mediated by humoral mechanisms arising from homeostatic T-cell proliferation.61 Absence of thyroid dysfunction in neoplastic conditions in comparison to MS, is presumed to be due to simultaneous use of other immunosuppressive agents in patients with cancer or to the tendency for underlying autoimmunity in MS patients.1
Thalidomide analogues
Thalidomide and its analogues lenalidomide and pomalidomide are used for treatment of multiple myeloma, myelodysplastic syndrome and mantle cell lymphoma. The anticancer effects are mediated by antiangiogenic, antiproliferative and immunomodulatory activity.62 The incidence of subclinical hypothyroidism is 20% and overt hypothyroidism is 7% during the first 6 months of thalidomide therapy.63 In a retrospective series of 170 patients receiving lenalidomide, hypothyroidism was reported in six, and thyrotoxicosis in four.64 In another series, lenalidomide caused hypothyroidism in 25.8% of cases after a median duration of 5.2 months in patients with diffuse large B-cell lymphoma.65 Two case reports of pomalidomide-induced hypothyroidism have been described in the literature.66,67 Slean and Silkiss reported a case of lenalidomide induced eyelid retraction in a 76 year old woman who was euthyroid but had elevated thyroid antibodies.68
One of the probable mechanisms behind development of hypothyroidism is induction of ischaemic thyroiditis through antiangiogenic effect of thalidomide. Deregulation of cytokine or direct effects on T-lymphocytes has also been hypothesised to trigger an autoimmune response to thyroid.63 Elevated anti-thyroid peroxidase antibodies and increased TNF-α levels have been demonstrated in these patients, further corroborating the possibility of immune-mediated pathogenesis.65,69 Additionally, lenalidomide might cause increased T-cell proliferation and activation by stimulating tyrosine phosphorylation of T-cell costimulatory molecules (e.g., CD28). This may inhibit regulatory T cells and might block immune checkpoints like CTLA-4.70,71 Measurement of TSH is recommended before initiation of treatment with these agents, and every 2–3 months thereafter for the duration of therapy.72
Radioiodine-based cancer therapy
Metaiodobenzylguanidine (MIBG) is a norepinephrine analogue. When combined with I-131, it is used as radiotherapeutic agent to treat tumours derived from neural crest cells. Active transport by uptake-1 system is the dominant mechanism of uptake in these cells, while passive diffusion plays a minor role.73
Indications and adverse effects
I-131 MIBG therapy has been used for a variety of tumours including inoperable pheochromocytoma or paraganglioma, stage III or IV neuroblastoma, inoperable carcinoid tumours, and metastatic or recurrent medullary thyroid carcinoma.74 Adequate uptake and retention of MIBG during a pre-therapy scan is a prerequisite before administration of a therapeutic dose of I-131 MIBG. The adverse effects associated with therapeutic use of I-131 MIBG include transient hypertension, bone marrow suppression and thyroid dysfunction.73 During its administration, free radioiodide formed due to chemical instability and biological degradation in liver, may result in toxicity to the thyroid gland.
Spectrum of thyroid dysfunction
Thyroid dysfunction following I-131 MIBG therapy has been reported in the form of hypothyroidism, radiation thyroiditis, thyroid nodule and thyroid carcinoma.75,76 Due to higher radio-sensitivity, the concern remains greater in children receiving this therapy, most commonly for neuroblastoma. Traditionally, when using Iodine-123 or I-131 MIBG for diagnostic or therapeutic purpose, thyroid protection is rendered using saturated solution of potassium iodide or Lugol’s iodine.77 Despite the use of thyroid protection, the incidence of thyroid dysfunction following MIBG therapy is quite high, and varies from 52–86% across studies.77–79
Incidence of thyroid dysfunction
In a study by van Santen et al., TSH elevation (>4.5 mIU/L) was reported in 22/42 (52%) subjects with neuroblastoma at a mean follow-up duration of 1.4 years following MIBG therapy.77 In another study by the same group, long-term thyroid outcomes among 25 neuroblastoma survivors were reported. After a median follow-up of 6 years, 14/25 (56%) subjects had persistent TSH elevation, while 6/25 (24%) developed thyroid nodule or cysts.78 In another study, by Picco et al., at a follow-up of 6–12 months following MIBG therapy, TSH elevation was evident in 12/14 (86%) neuroblastoma survivors. Of these, eight (67%) had overt hypothyroidism, while four (33%) had compensated hypothyroidism.79 In a study comparing two thyroid protection strategies, a combination of potassium iodide, thyroxine and methimazole (cocktail protection) was compared with potassium iodide (standard protection) in children with neuroblastoma receiving therapeutic I-131 MIBG. The study evaluated 23 children who were given the cocktail protection (beginning 1 day before diagnostic I-131 MIBG and continued till 4 weeks after therapeutic I-131 MIBG), and were followed up for a mean duration of 19 months. The control group was historical cohort of children with neuroblastoma (n=42) who received standard thyroid protection. Thyroid function was normal in 86% survivors in group A (cocktail protection) compared to 44% in group B (standard protection). The authors thus concluded, that a combined protective strategy may be more effective against radiation damage in patients receiving MIBG therapy for neuroblastoma.80 The long-term efficacy of this strategy, however, remains to be seen.
Screening and management
Although a clear recommendation is lacking, it is advisable to obtain a thyroid function at baseline and at every 3–6 monthly interval in patients receiving therapeutic I-131 MIBG. LT4 supplementation is recommended in those with low T4 levels or TSH level >10 mIU/L. However, considering the theoretical concern of increased neoplasm risk with chronically elevated TSH levels, some authors recommend treating even mild subclinical hypothyroidism (TSH <10 mIU/L) in the setting of previous radiation exposure.81 Since thyroid radiation is a known risk factor for malignancy, and increased incidence of thyroid nodule has been reported in studies, such patients should be followed indefinitely for the same.76,78
Bexarotene
Topical and systemic preparation of bexarotene, a third-generation retinoid analogue, is approved for treatment of cutaneous T-cell lymphoma. It is a selective agonist of retinoid X receptor (RXR), a member of the nuclear receptor superfamily. RXR functions by forming heterodimer with thyroid hormone receptor and other nuclear receptors. T3 exerts a negative feedback effect on transcription of the β-subunit of TSH after binding to its receptor and subsequent heterodimerisation with RXR.
Mechanism of thyroid dysfunction
Bexarotene alters the feedback effect of thyroid hormone on pituitary by causing reversible thyroid hormone independent inhibition of TSH gene expression. It blocks transcription even in the absence of T3 and decreases TSH production.82,83 Besides, bexarotene causes direct inhibition of TSH secretion and rapid fall in serum TSH occurs even after administration of a single dose.84
Central hypothyroidism
Oral bexarotene administration has been found to induce rapid onset and reversible central hypothyroidism.82 The percentage of patients developing central hypothyroidism, in trials with more than 20 patients on bexarotene, varied from 29–100%.82,85–92 In a recently published study from Japan, hypothyroidism developed at 1 week in 45 out of 66 patients and only five patients remained euthyroid at 1 month.92 Serum TSH levels returned to normal within a week of discontinuation of bexarotene in 90% of cases who were euthyroid earlier.82
The factors predicting the development of central hypothyroidism are higher doses of bexarotene (>300 mg/m2/day) and prior treatment with IFN-α.82 The aforementioned Japanese study also demonstrated that, a pre-treatment FT4 value in the lower–normal range, or a pre-treatment TSH value <1.30 mIU/L, correlated with development of central hypothyroidism. Iodine deficiency and polymorphism of RXRγ1 gene were also proposed as other possible determinants of development of central hypothyroidism.92 RXRγ and negative thyroid hormone responsive element are both present in the promoter region of the gene encoding the TSH-β subunit, and binds to retinoid and thyroid receptor respectively to inhibit its transcription.93
Other effects on thyroid metabolism
Bexarotene also exerts peripheral effects by increasing degradation of thyroid hormones. Ten post-thyroidectomy patients with pulmonary metastases of differentiated thyroid carcinoma who received a 6-week redifferentiation course of bexarotene, had a drastic fall in serum total T4, FT4 and total T3 levels. The increased catabolism of thyroid hormone was thought to be independent of peripheral deiodinase activity, and likely to be mediated by other pathways like glucuronidation and sulfation.94
Management
The recommendations suggest initiation of LT4 at 25–50 µg per day when bexarotene treatment is started.95–97 FT4 levels should be monitored every 1–2 weeks for first 4 weeks and adjusted to maintain FT4 within the upper third of the local laboratory normal range. There is no role of monitoring serum TSH levels. Once two consecutive blood reports are stable, frequency of monitoring can be decreased to once every 4 weeks. LT4 should be discontinued on stopping bexarotene unless the individuals have pre-existing primary hypothyroidism. Those with pre-existing primary hypothyroidism will have an increased LT4 requirement as bexarotene increases peripheral degradation of thyroid hormones. The dose of LT4 should be adjusted according to T4 levels and the dose requirement can increase two to three times. On stopping bexarotene, the dose of LT4 should be reduced to pre-treatment dose. Maintaining normal thyroid function has a beneficial effect on clearance of lipids and will help in the management of hypertriglyceridemia, another known complication of bexarotene.
Conclusion
Immune-based and targeted chemotherapy agents can produce a distinct group of immune-related adverse events, foremost among them being thyroid dysfunction. Appropriate screening for primary thyroid dysfunction should be performed when immune checkpoint inhibitors, TKI, IFN-α, IL-2, thalidomide and other immunomodulatory drugs are administered. The guidelines and recommendations on management of thyroid disorders induced by common anticancer drugs are summarised in Table 2. Central hypothyroidism is also described as a common adverse event with bexarotene and immune checkpoint inhibitors, albeit through different mechanisms. Administration of LT4 should be instituted when appropriate. In many cases the thyroid dysfunction is temporary and treatment can be withdrawn with discontinuation of anticancer therapy.