




The clinical presentation of hypocalcemia varies based on the degree and chronicity of the derangement. Although many patients remain asymptomatic, mild symptoms of hypocalcemia include circumoral and extremity paresthesia, Chvostek’s sign and Trousseau’s sign; more severe derangements can present with cardiac arrythmias and seizures.1–3 Chronic hypocalcemia may present with cataracts and basal ganglia calcifications.2,3 Due to the wide variability of clinical presentation, it is imperative to have a low threshold of suspicion for hypocalcemia. Initial work-up includes laboratory evaluation of serum creatinine, magnesium, vitamin D, phosphate, and parathyroid hormone (PTH).2,3 Although rare, in the setting of hypocalcemia, hyperphosphatemia, and elevated PTH, pseudohypoparathyroidism (PHP) is an important diagnosis to consider.
PHP results from end-organ resistance to PTH, and manifests primarily with symptoms of hypocalcemia.2,4 The resistance is mediated by genetic mutations in the coding and regulatory regions of GNAS, a gene encoding the alpha-subunit of the G-protein that mediates PTH and other hormone activity via production of second messengers.4–6 Clinical and biochemical data have been used to subcategorize the disease into types 1a, 1b, 1c, and 2.3,4,6 The presence of Albright hereditary osteodystrophy (AHO), a constellation of somatic features including brachydactyly, round facies, obesity, short stature and ectopic calcifications,2–4,6,7 has classically ruled out PHP1b and ruled in PHP1a. In this case report, we present a female with phenotypical features of AHO but genetic mutations consistent with PHP1b. We also highlight the neuromuscular irritability seen with hypocalcemia, including laryngospasm.
Case description
A 16-year-old Caucasian female presented to an outside hospital with symptoms of bilateral paresthesias and cramping in her hands. She denied previous episodes. She had recently been evaluated for ongoing productive cough with dyspnea of 3 weeks’ duration; all diagnostic tests were negative, and she was treated with corticosteroids and antibiotics for presumptive pneumonia, but symptoms did not resolve. The patient’s past medical history was significant for premature birth at 29 weeks’ gestation, and twin gestation. Family history was negative for metabolic, renal, and calcium disorders. Of note, the family reported that the patient and her maternal grandmother shared a distinctive phenotype with round facies and short stature. She reported regular menstrual cycles since menarche at age 11 or 12. She denied alcohol, tobacco, or illicit drug use, and was not taking any additional medications or supplements.
Admission laboratory test results at an outside hospital showed decreased calcium and 25-hydroxyvitamin D, elevated phosphate and PTH, and normal-range magnesium and creatinine (Table 1, Column A). She received a single dose of vitamin D 50,000 international units (IU) and calcium gluconate intravenously (IV), and her paresthesias resolved. She was started on calcium carbonate salt 150 mg by mouth three times daily. On day 3 of admission, she developed bradycardia to 30 bpm and a 4-second episode of ectopy on electrocardiogram. She received one dose of calcium gluconate IV and magnesium sulphate IV, and was transferred to our tertiary care level pediatric hospital.
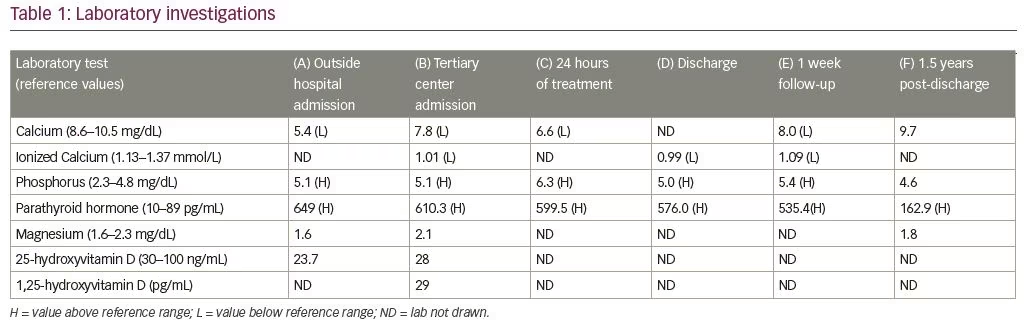
At the time of transfer, the patient’s vital signs were stable, and laboratory tests were ordered (Table 1, Column B). Physical examination was significant for round facies, 2+ reflexes, and productive cough, and was negative for Chvostek’s sign, Trousseau’s sign, and clonus. The patient was placed on cardio-respiratory monitoring, and nephrology and endocrinology specialists were consulted.
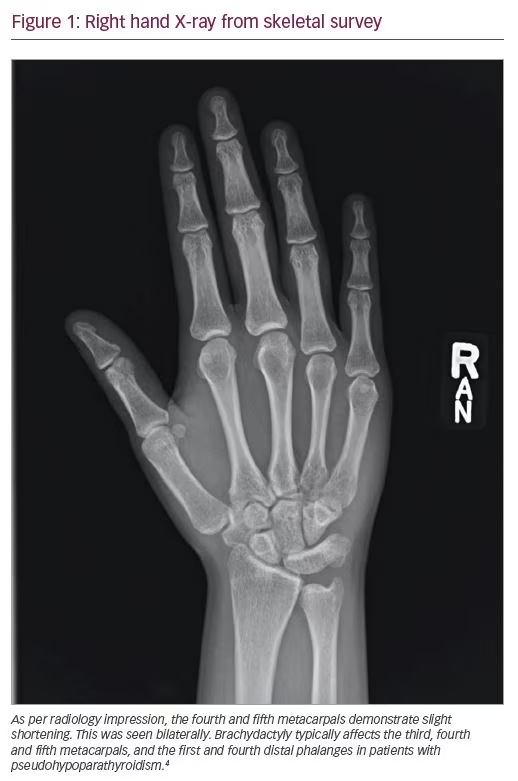
Differential diagnoses included vitamin D deficiency, renal anomalies, and PTH resistance. The patient’s 25-hydroxyvitamin D and 1,25-hydroxyvitamin D levels returned marginally below normal (Table 1, Column B); thus, suspicion remained low for vitamin D deficiency as the cause of hypocalcemia. Renal disease was unlikely due to normal creatinine values and normal renal ultrasound findings. Urine studies did not demonstrate calcium-wasting disease. PHP remained high on the differential. Differentiating PHP1a from PHP1b remained difficult clinically: she had features of AHO, including round facies, short stature (below third percentile), and shortening of the fourth and fifth metacarpals on skeletal survey imaging (Figure 1), all of which supported a diagnosis of PHP1a. However, her insulin growth factor 1, luteinizing hormone, and follicle-stimulating hormone levels did not indicate other end-organ resistance endocrinopathies, which is often seen in PHP1a. Thyroid function tests showed low–normal free T4 (0.8 ng/dL) and normal thyroid-stimulating hormone (TSH) (2.76 mcIU/mL), possibly indicating mild resistance. Bone X-rays were negative for rickets and osteitis fibrosa, and demonstrated a bone age of 17 years. While genetic testing was pending, the patient was started on calcium carbonate salt 1,250 mg four times daily, and daily calcitriol 0.5 mcg. Calcium, ionized calcium, phosphate, and PTH were monitored multiple times daily throughout the patient’s 5-day hospitalization (Table 1, Column C). With this treatment alone, her cough completely resolved in a few days. An additional calcitriol 0.25 mcg dose was added to further decrease PTH levels, and mild improvement was seen in the laboratory value at time of discharge (Table 1, Column D).
Cytogenic testing showed complete loss of methylation at exon 1A of the GNAS complex locus (Johns Hopkins DNA Diagnostic Laboratory), consistent with a genetic diagnosis of PHP1b. No variants were detected within the coding regions or splice junctions of the GNAS gene. The patient was followed in the endocrinology clinic for 1 week post-discharge (Table 1, Column E). She developed autoimmune thyroid disease 11 months after initial presentation (TSH 52 mcIU/mL, free T4 0.4 ng/dL, thyroid peroxidase antibody 2,397 IU, thyroid globulin antibody 103 IU), and responded to thyroid replacement treatment. Her calcium and calcitriol supplements were titrated as needed for the next 1.5 years, at which time she was lost to follow-up. Her last laboratory results are detailed in Table 1, Column F.
Discussion
Classification of PHP has been difficult, because prior paradigms emphasized categorization based on the presence or absence of AHO. However, in recent years, it has become more apparent that there is a wide variability of phenotypes with each genotype.4,6 PHP1a is a result of mutations within the coding regions of GNAS, and presents with PTH and other end-organ resistance and AHO.2–4,6 PHP1b is a result of mutations within regulatory regions of GNAS, and presents with PTH resistance but without AHO.3,4,6–8 There are four distinct, differentially methylated regions (DMRs) that can be implicated in PHP1b: GNAS-NESP:TSS-DMR, GNAS-AS1:TSS-DMR, GNAS-XL:Ex1-DMR, and GNAS A/B:TSS-DMR.4,9 Patients may have involvement of one or multiple DMR sites resulting in disease, but loss of methylation at GNAS A/B:TSS-DMR, also known as exon 1A, is implicated in all cases.4,8–10 Our patient had complete loss of methylation at exon1A, which has been postulated to be a genetic silencer; therefore, loss of methylation results in expression of the silencer, and decreased expression of GNAS.11 Unfortunately, the genetic testing report did not explicitly describe the state of the other three DMRs of the GNAS locus; this information is important in classifying disease and determining inheritance patterns.9 Furthermore, some guidelines suggest analyzing STX16 for deletions and duplications in patients with methylation defects isolated to exon 1A.10 Additionally, the patient was lost to follow-up, inhibiting further genetic testing of her family members. Nevertheless, her genetic studies supported a diagnosis of PHP1b; however, clinically, she had AHO, supporting PHP1a. To our knowledge, fewer than 40 cases demonstrating this overlap exist in the literature.7,12
Our case demonstrates that the presence or absence of AHO cannot be used to differentiate between PHP1a and PHP1b, as traditional classification systems suggested.4,6,8 In 2018, an international consensus, developed by a panel of 37 experts in PHP, devised a new classification system emphasizing clinical and genetic testing in the subtyping of disease.4 Major criteria for the clinical diagnosis of PHP include evidence of PTH resistance, and/or AHO, and/or ectopic ossifications, and/or obesity before 2 years of age with TSH resistance.4 Minor criteria include endocrinopathies, such as hypothyroidism and growth hormone deficiency; neuropathies, including cognitive impairment and central nervous system (CNS) structural abnormalities; mineralization defects, evident by abnormalities of tooth enamel or CNS calcifications; and findings of sleep apnea, asthma, or early-onset obesity.4,9 Guidelines suggest completing genetic testing in all patients with one or more of the major criteria for PHP. If clinical suspicion exists for an abnormality of a specific gene, Sanger sequencing of the gene can be used; in cases where pathology of a specific gene is not obvious, a panel analyzing multiple proteins in the PTH signalling pathway can be utilized.4 Testing should include analysis of the DNA sequence, methylation pattern, and copy number variant at the GNAS locus, and parental testing is indicated if abnormalities are present.4,9 This case supports the use of this new system, as dependence on clinical features can be confusing due to overlapping phenotypes.
Proper subclassification of disease remains imperative, as different subtypes are associated with different comorbidities that the physician should be alert to when managing such cases. For example, TSH resistance is associated with all cases of PHP1a but only some cases of PHP1b.4 With knowledge of the subtype of disease, physicians can better understand prevalence of concomitant diseases, which can serve as important factors in clinical decision making and screening. Additionally, more appropriate genetic counselling can be offered to patients.
Mild degrees of TSH resistance in PHP1b have been described in a handful of cases.4,11,12 It is unclear if our patient’s borderline TSH represents mild resistance or her normal physiology. She later developed overt autoimmune mediated thyroiditis; however, the current literature does not provide a mechanism for this or emphasize an association between PHP and autoimmune thyroiditis. Of note, one case also reports development of autoimmune thyroid disease similar to our patient.12 An association between PHP and autoimmune thyroiditis may serve as a point of research in the future. Nevertheless, although the pathologies are not clearly linked, this case emphasizes the importance of continued surveillance for other endocrinopathies after diagnosis of PHP.
The patient’s productive cough and dyspnea resolved within days of treatment of her hypocalcemia. Clinical suspicion for pneumonia remained low because she had completed appropriate therapy with antibiotics and steroids without resolution of symptoms and the patient’s chest X-ray was reported as normal from the outside hospital. We suspect, from the clinical picture, that these symptoms were secondary to hypocalcemia-induced laryngospasm because of the correlation of symptom resolution with correction of her hypocalcemia. Although no clear mechanism links hypocalcemia with productive cough, we believe that her dyspnea resulted from neuromuscular irritability of the larynx, and this hypersensitivity may have also triggered a cough reflex. Laryngospasm is an uncommon presentation of hypocalcemia, as severe derangements often produce more obvious abnormalities, such as seizures.2,3 When it does occur, laryngospasm typically presents with symptoms of acute airway compromise, including stridor and severe dyspnea.1,13 Our patient did not present with such concerning respiratory symptoms, but instead with a more stable, chronic, indolent course. Another case in the literature describes a teenager who presented after a 2-day course of intermittent dysphagia, stridor, and episodes of gasping for air due to hypocalcemia from PHP1b.14 This case, and our own, highlight that upper airway distress from hypocalcemia can present in a plethora of manners, and not just with severe symptoms.
In conclusion, we present a case of PHP1b that supports the use of newer classification systems that emphasize genetic testing, not clinical phenotype, as the most important data in classification of PHP. Additionally, this case demonstrates hypocalcemia-induced laryngospasm, contributing to the literature of a rare symptom of this electrolyte disorder.
Limitations
The patient’s genetic report did not specify the state of the three other DMRs implicated in disease, which limits understanding of inheritance. Additionally, genetic analysis was not conducted in her family members. Laryngoscopy was not completed, but may have helped confirm laryngospasm as the cause of her dyspnea and cough. Finally, patient was lost to follow-up, inhibiting the authors from obtaining consent. Detail has been removed from this case description to ensure anonymity.


