Phaeochromocytomas/paragangliomas (PHAEO/PG) are chromaffin cell tumours that can present sporadically or as part of other familial syndromes including hereditary paraganglioma syndromes, multiple endocrine neoplasia 2, von Hippel-Lindau syndrome and neurofibromatosis type 1 (NF1). PHAEO/PG are found in about 0.2% of patients with hypertension1 and in 5% of all cases of adrenal incidentalomas.2,3 The vast majority of PHAEO/PG produce catecholamines, which are metabolised into metanephrines and 3-MT (methoxytyramine). PHAEO/PG are treated with surgical resection followed by lifelong biochemical and radiological screening, especially in those patients at high risk of recurrent and/or metastatic disease.4
Although these tumours can occur at any age, they are more prevalent during the fourth to fifth decades5 with a diverse clinical presentation that can range from being asymptomatically discovered (incidental), to the typical attacks, such as paroxysmal headache, sweating, palpitations/tachycardia,1,6 and patients may also present with fatal complications. Indeed, many patients do not have the classic triad of symptoms7,8 and around 5–15% of patients can be normotensive.6,9,10
NF1 is an autosomal dominant neuro-cutaneous disorder with an estimated birth incidence of 1 in every 2,500–3,000 individuals.11 The clinical criteria are robust12 and by using RNA-based assays, mutations in the NF1 can be detected in >95% of those meeting the clinical criteria.13 While it is recommended to screen for PHAEO/PG in individuals with other predisposing genetic disorders, neither adult nor paediatric NF1 guidelines recommend routine biochemical screening in patients with NF1 unless they are hypertensive or symptomatic.14,15 Patients with undiagnosed PHAEO/PG are at risk of developing life-threatening cardiovascular complications due to PHAEO crises triggered by tumour manipulation, anaesthesia, drugs, pregnancy,16 or rarely, metastatic disease. Over the last few years, there has been increased attention in the literature to the potential significance and need for biochemical screening in patients with NF1 for PHAEO/PG. As asymptomatic and normotensive patients with NF1 are not suitable candidates for screening as per the currently practiced recommendations, the aim of this study was to examine the safety and impact of such approach on patients with NF1 who ultimately present with PHAEO/PG, by looking into the mode of presentation and the incidence of any adverse outcomes.
Methods
Adult patients with a diagnosis of NF1 and PHAEO/PG were identified via a retrospective search of hospital records from a tertiary referral unit in the north-east of England (The Royal Victoria Infirmary – Newcastle Hospitals NHS Foundation Trust) from 1998–2018. Caldicott approval was obtained prior to data collection. Retrospectively collected data for all resected PHAEO/PG was obtained and matched with a computer-based search for all histologically confirmed PHAEO/PG. Using this approach, it was possible to track those cases of NF1 with PHAEO/PG managed at different hospitals within the region. A parallel computer search on the NF1 register database was performed by the national complex NF1 service based at Manchester Centre of Genomic Medicine.
A diagnosis of NF1 identified by the database search was confirmed by reviewing the clinical records complying with the clinical criteria of NF1 and/or genetic testing. The diagnosis of PHAEO/PG was based on biochemical, radiological and histological criteria. A diagnosis of hypertension was assigned to patients stated to have such a diagnosis or described as being on anti-hypertensive medications. Classical PHAEO symptoms were defined as the presence of any of the following: headaches, palpitations and sweating.1,6,17 Review of medical records, including history and examination, was performed retrospectively to identify the presence of any classical symptoms, even in those patients where the diagnosis was found incidentally.
Currently, Liquid chromotography with tandem mass spectrometry assay for measuring plasma metanephrines has been in use since 2010. Prior to this, plasma catecholamines were measured. For urinary metanephrines, liquid chromatography with electrochemical detection method has been in use since at least 2002. Because of the difference in the assays used in other hospitals across the north of England, and to account for the change in the preference for the assays used over time, the interpretations of the biochemical results were unified by adopting the use of a standard score.18 The standard score can be calculated as below:
Standard score = (current − highest value in the normal range)/highest value in the normal range
Patients’ demographics; mode of clinical presentation; biochemical, radiological and histology results; and the occurrence of death, metastases and any adverse complications attributed to hypertensive crisis or circulating catecholamines, were recorded.
Statistical analysis
Analysis of data was carried out using the available statistical package of SPSS-24 (Statistical Packages for Social Sciences version 24). The significance of the difference of different percentages (qualitative data) was tested using Pearson chi-square test (χ2-test) with the application of Yate’s correction or Fisher’s exact test whenever applicable. Statistical significance was considered whenever the p-value was ≤0.05.
Results
Patients demographics
Of 180 patients who had PHAEO/PG resection and 3,715 with NF1,
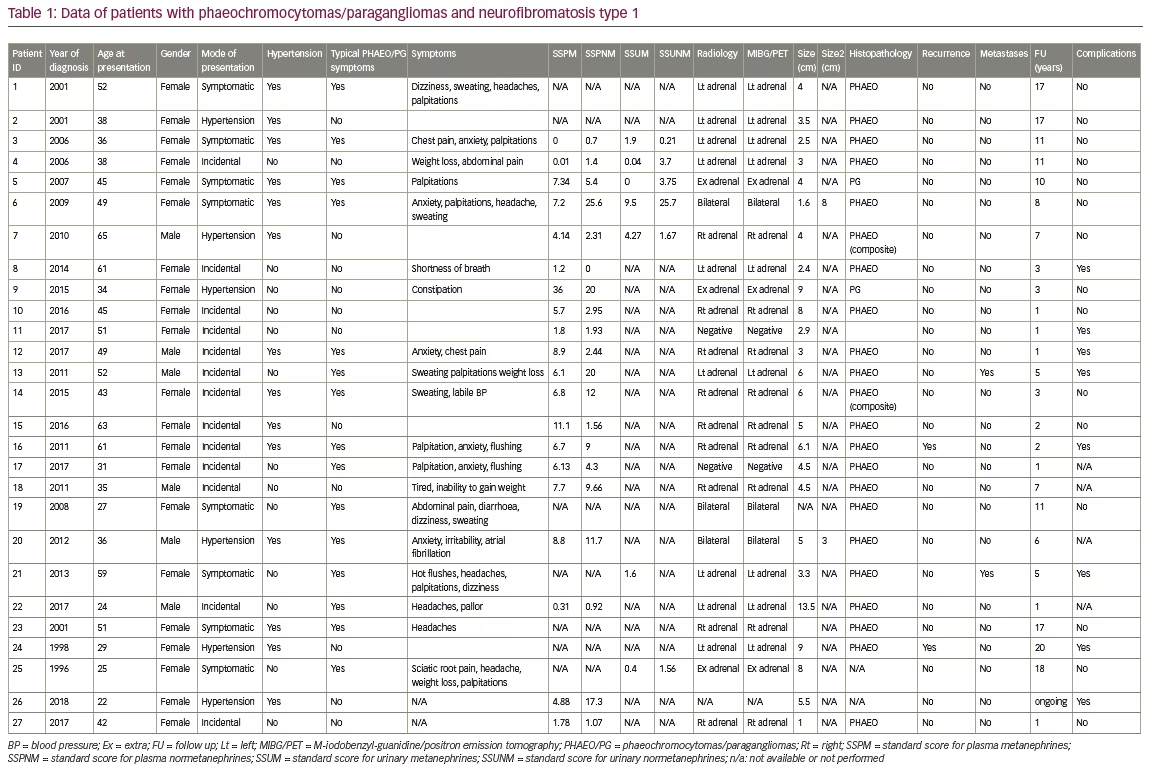
27 patients with dual diagnosis of NF1 and PHAEO/PG were identified (Table 1). Fifteen of these patients were identified by searching the hospital records with further 12 patients were identified through the NF1 database. All patients had a clinical diagnosis of NF1 before PHAEO was diagnosed except for one patient, in whom genetic testing after PHAEO resection confirmed a pathological NF1 mutation. Two patients were siblings. The mean age at presentation with PHAEO was 43.1 ± 12.7 years (range 22–65) and 78% (n=21) were females.
Mode of presentation
The mode of presentation was incidental (tumours were identified by imaging performed for other indications) in 48% (n=13) of patients, 30% (n=8) of patients presented with symptoms, and 22% (n=6) were identified during investigations for secondary hypertension. Typical PHAEO symptoms were reported in 55% (n=15) (including those patients where the diagnosis was found incidentally) and 52% (n=14) were hypertensive prior to PHAEO/PG diagnosis. No patient had undergone biochemical screening for PHAEO/PG before the later diagnosis was evident.
Diagnosis confirmation
Imaging confirmed the presence of a unilateral adrenal mass in 78% (n=21) of patients, a bilateral adrenal mass in 11% (n=3), and an extra-adrenal location in 11% (n=3). Mean tumour size was 5.0 ± 2.8 cm (range 1.0–13.5). Biochemical results were availeable in 23/27 patients. All patients had plasma and/or urinary catecholamines elevated with mean standard score for plasma metanephrines (n=20) of 6.6 ± 7.6 (range 0–36), plasma normetanephrines (n=20) 7.5 ± 7.8 (range 0.0–25.6), urinary metanephrines (n=7) 7.5 ± 7.8 (range 0.0–25.6), and urinary normetanephrines (n=6) 6.1 ± 9.7 (range 0.21–25.7). Histology results were available for 23 patients. Adrenal PHAEO was confirmed in
21 patients (of which, two patients had composite PHAEO-ganglioneuroma). Paraganglioma was confirmed histologically in two patients. Another patient with a pelvic neurofibroma was assumed to have intra-fibroma paraganglioma tissue as metanephrines levels normalised after neurofibroma resection. All patients received medical therapy with an appropriate alpha-blockade agent prior to consideration of surgery.
Adverse clinical outcomes
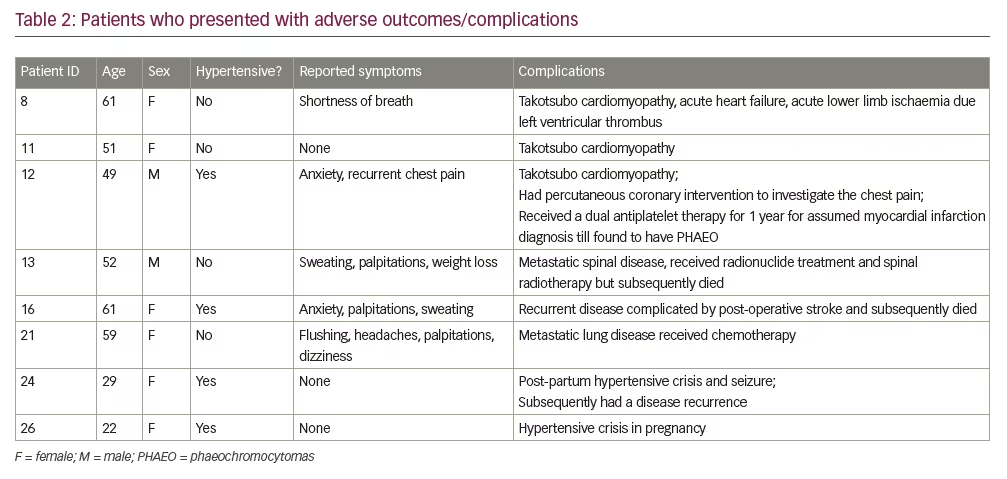
Potentially avoidable complications had occurred in 8/27 of patients at the time of diagnosis of PHAEO/PG (Table 2). Among these eight patients with documented complications, four were hypertensive and only five presented with classical PHAEO/PG related symptoms. Three patients were found to have Takotsubo cardiomyopathy including one patient who presented with acute heart failure and acute lower limb ischemia due to left ventricular thrombus formation needing embolectomy. In another patient there was a 7-year history of anxiety, low mood, and chest pain. The patient received a diagnosis of non-ST elevation myocardial infarction and treated with dual anti-platelet therapy and ß receptors blockers for 1 year before subsequent cardiac catheterisation and magnetic resonance imaging showed no evidence of ischemic heart disease. Indeed, the delay in recognising PHAEO in this patient resulted in exposure to the risks of an invasive cardiac procedure as well as the risk of worsening hypertension and bleeding due to the use of ß blockers and dual anti-platelet therapy.
Two patients had metastatic disease; one patient received surgical resection followed by chemotherapy while the other patient presented with a fall and multiple fractures with subsequent diagnosis of
metastatic PHAEO that was palliatively treated with radionuclide therapy before his death.
Two patients had disease recurrence. Another patient died as a result of post-adrenalectomy stroke and two young females suffered hypertensive crises during pregnancy/after delivery. As compared to patients who did not present with complications, there was no significant statistical difference in age (p=0.389), sex (p=0.214), tumour size (p=0.863), presence of hypertension (p=0.645), presence of typical PHAEO symptoms (p=0.879), or catecholamines levels (p=0.498).
Prevalence of PHAEO/PG in NF1
The prevalence of PHAEO/PG in patients with NF1 remains debateable. While the mostly cited prevalence in the literature is 0.1–5.7% based on a retrospective review in 1999 of families with NF1,19 subsequent studies showed that the prevalence might be higher if patients were screened prospectively. In a prospective study of 156 patients with NF1, the prevalence of PHEO/PG in NF1 was 7.7%.20 An even higher prevalence (14.6%) was reported when 48 consecutive patients with NF1 were screened prospectively.21 This indicates that the association between NF1 and PHAEO/PG could have been underestimated in the past.
Mode of Presentation
In the last few years, an increase in the number of patients with PHAEO/PG detected incidentally was evident in those who are normotensive and asymptomatic given the widespread use of advanced imaging modalities and a better understanding of the genetic basis of the disease.2,3,8,18,22, In the cohort presented here, PHAEO/PG was discovered incidentally in 48% of subjects with NF1 with a significant percentage of patients diagnosed in the absence of classical symptoms or elevated blood pressure.
While sustained and/or paroxysmal hypertension is a feature of PHAEO in 80–90% of affected individuals, 5–15% of patients could be normotensive at diagnosis.23 Even with increased circulating catecholamines levels, blood pressure may not be elevated due to multiple factors including catecholamines-induced hypovolemia due to vasoconstriction, the rate of catecholamines inactivation and availability to target cells, the reactivity of vascular smooth muscle, and catecholamines receptor numbers and sensitivity.24 It has been found that chronic exposure to a high level of catecholamines may result in desensitisation of the postjunctional α1- and α2-adrenergic receptor causing an attenuation to their vasopressor responses.25 In the present study, 14 out of 27 patients (52%) were not known to be hypertensive before the diagnosis of PHAEO/PG.
The classical triad of paroxysmal headache, palpitations and sweating is strongly associated with catecholamines excess and often described as ‘an attack’.17 However, the disease is still referred to as ‘the great mimic’ as the clinical presentation can be diverse with non-specific signs and symptoms.26 In the present study, 15 out of 27 patients (56%) presented with at least one of the classical symptoms while the remaining patients presented with a variety of other symptoms (Table 1).
Review of the literature
Several studies in the literature have reported heterogeneity in the presentation of PHAEO/PG in NF1 (Table 318,20,21,27–31) and questioned the significance of screening.
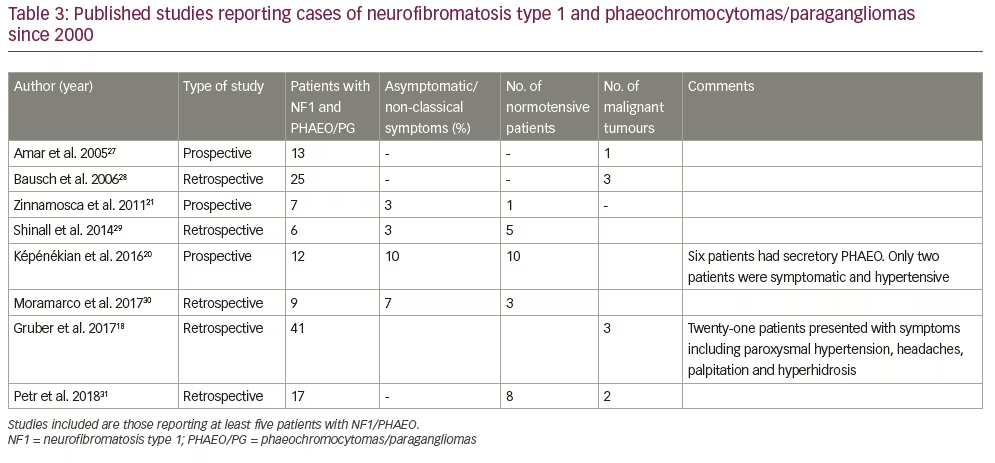
In a large retrospective study of PHAEO/PG in NF1 population, Gruber et al. studied 41 patients from the Mayo clinic database between 1959–2015 and found that the diagnosis of PHAEO/PG was mostly incidental over the last 2 decades, with only three patients (7.3%) who were found through biochemical case detection.18 Moreover, the authors observed that all cases of bilateral, recurrent and metastatic diseases occurred in women. In their study, the authors recommended biochemical screening for PHAEO/PG in all patients with NF1 at an early age (age 10–14 years) with a repeat evaluation every 3 years and before elective surgical interventions and conception.
In another study, Képénékian et al. prospectively screened 156 patients with NF1 using a combination of urinary metanephrines and abdominal imaging and subsequently found 12 cases of PHAEO/PG.20 Without this prospective methodology, only two patients out of the 12, could have been diagnosed using the current approach of electing only symptomatic and hypertensive patients for screening. In this study, the authors suggested initiating screening every 4–5 years after 40 years of age.
Furthermore, significant adverse outcomes have been described in a cohort of NF1 patients who were ultimately found to have PHAEO/PG. Petr et al. recently studied 17 patients with NF1/PHAEO/PG; two patients were found to have metastatic disease and six suffered cardiovascular crises, especially during surgical procedures and/or delivery.31 Butz et al. investigated the peri-operative outcomes of PHAEO/PG resection in a cohort of patients at the Mayo clinic from 2000–2016 and found that patients with NF1 suffer the worse intraoperative hemodynamic course and more severe postoperative complications, as compared to other PHAEO/PG genetic syndromes.32 This was felt due to larger tumour size and higher levels of catecholamines as compared to other familial PHAEO/PG syndromes where more strict and timely screening is applied.
A recent review of all published isolated case reports of PHAEO/PG in patients with NF1 found at least 73 cases; 36/73 did not present with any of classical symptoms while 27/73 were normotensive at the time of PHAEO/PG diagnosis.33 Furthermore, it was found that 31/73 of patients presented with life-threatening complications and interestingly, the majority were relatively young (age <40 years). These findings, in contrast to Képénékian et al.,20 would favour considering screening at an earlier age.
Current practice
The currently practiced guidelines for the management of NF1 recommend that patients with NF1 should have a specialist clinic visit once a year where clinical evaluation is performed and blood pressure is measured given the association with renal artery stenosis and PHAEO.14 According to the American College of Medical Genetics and Genomics, only patients with NF1 and hypertension, aged ≥30 years, who are pregnant, and/or symptomatic should be considered for biochemical or imaging screening given the lack of any randomised studies on the efficacy of biochemical or imaging screening to detect PHAEO in asymptomatic patients with NF1.34 The presumed perception of an infrequent occurrence of PHAEO/PG in NF1 could also be another reason for the lack of an agreed consensus with regards to screening asymptomatic patients.
However, the value of screening needs to be reconsidered in light of the emerging findings from retrospective and prospective studies over the last few years. The currently perceived low risk of PHAEO/PG in NF1 is likely to be underestimated as most of our knowledge is derived from retrospective case series with a small number of patients, albeit higher prevalence was evident on prospective case detection.20,21 In addition, it is clear that the current case detection strategy fails to identify those patients who are incidentally confirmed to have PHAEO/PG. Finally, the observations presented in the current study, in addition to the evidence provided from recent studies,27,28 suggest that NF1 subjects are at risk of developing catastrophic sequalae upon under-recognition of PHAEO/PG.
The literature and these findings suggest that majority of patients with NF1 who were ultimately found to have PHAEO/PG, reported non-specific symptoms and had been through multiple consultations, extensive investigations, and invasive procedures over the years before the diagnosis of PHAEO/PG was reached. Besides the risk of catastrophic complications that may eventually arise, this patient pathway generates additional medical costs, hospital visits and uncertainty, which negatively impacting the patient’s mental and physical wellbeing. In view of the above, it may be argued that routine screening could be safe and cost-effective in reducing the burden, morbidity and mortality in patients with NF1. A well-designed randomised controlled trial or a population based-study is necessary to address this important question.
Limitations and strengths
This study is retrospective and therefore the strength of evidence is relatively low. The NF1 register may not be representative of the true prevalence of NF1, a potential source of bias. Also, it was challenging to track all the medical records and obtain full clinical information as many patients were treated in different units across northern England. Despite those caveats, to our knowledge, this series represents the largest European cohort of NF1 and PHAEO/PG and one of the few to look into the adverse clinical outcomes over the last 20 years.
Conclusion
Relying on symptoms and blood pressure status to decide if screening for PHAEO/PG is needed in patients with NF1 may be associated with unnecessary risk and avoidable harm. We recommend measuring urinary or plasma metanephrines at an early age (10–14 years) and repeating those measurements every 3 years in all patients with NF1 regardless of the way they present. Such an approach is less periodic as compared to other hereditary PHAEO/PG syndromes where frequent surveillance is needed and this can be conceptualised in light of the lower prevalence, penetrance rate and incidence of multifocal disease in NF1.32,34–36 Subsequent prospective studies examining and comparing the clinical outcomes of routine biochemical screening versus the current surveillance practice in the NF1 population are warranted.