Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant endocrine tumour syndrome characterised by three main manifestations which are primary hyperparathyroidism (78–94%), gastroenteropancreatic neuroendocrine tumours (GEP-NETs) (35–78%) and pituitary adenomas (20–65%).1 As described, GEP-NETs are the second most common component of MEN1 syndrome and metastatic GEP-NETs are shown to be related with poor prognosis.2
For metastatic and inoperable GEP-NETs, there are some interventional and medical therapies. Peptide receptor radionuclide therapy (PRRT) with Yttrium-90 (90Y) and Lutetium-177 (177Lu) is one of the important radiotherapies; however, there have been no randomised controlled study evaluating both efficacy and safety of 177Lu-DOTATATE since 2017. The recently published NETTER-1 study demonstrated the markedly longer progression-free survival (PFS) with 177Lu-DOTATATE treatment in midgut NETs.3
In this case report, we describe a patient with MEN1 syndrome with inoperable metastatic GEP-NETs who had excellent response to the treatment with six cycles of 177Lu-DOTATATE.
Case report
A 35-year-old male patient was admitted to our clinic with widening of hands and feet, polyuria, polydipsia, nausea, vomiting and constipation in 2008. He had a history of hydrocephalus diagnosis when 2 months old. Physical examination showed normal vital signs and findings, except coarsening of voice and large tongue, nose, hands and feet. Laboratory findings revealed low total testosterone (0.14 ng/mL), luteinising hormone (0.66 mIU/mL) and follicle stimulating hormone (2.24 mIU/mL); elevated Insulin like growth factor-1 (IGF-1) (662 ng/mL; according to age- and sex-specific serum IGF-1 levels), growth hormone (GH) (22.7 ng/mL), and calcium (11.0 mg/dl); and intact parathyroid hormone concentration (160 pg/mL) with low phosphate concentration (2.1 mg/dl). Twenty-four-hour urinary calcium excretion was elevated (399 mg/day). Additionally, bone mineral density was also decreased; L1–L4 anteroposterior spine T- and Z-scores were -3.4 SD
and -4.2 SD, respectively. Scanning with Technetium-99m-hexakis 2-methoxyisobutyi isonitrile sesta (99mTc-MIBI) demonstrated a focal area of increased activity at the left thyroid lobe inferior-pole region, suggestive of a parathyroid adenoma. These findings, including non-suppressed GH levels by oral glucose tolerance test (OGTT), were compatible with primary hyperparathyroidism, acromegaly and secondary hypogonadism; as a result, he was diagnosed with MEN1 syndrome.
He was also diagnosed with central diabetes insipidus by an overnight water deprivation test.
Pituitary magnetic resonance imaging (MRI) revealed a 19 mm pituitary macroadenoma without compression of optic chiasm and he was operated on by transsphenoidal route. Immunhistochemically, the pituitary adenoma was strongly and diffuse stained with GH, focal slightly stained with prolactin. Adrenocorticotrophic hormone staining was not detected. Ki67 index was approximately 8%.
At the patient’s follow-up evaluation after surgery, IGF-1 level was still elevated and there was no GH suppression after OGTT. Lanreotide 120 mg/month and cabergoline treatments were initiated together with desmopressin, and testosterone. Because of the presence of MEN1 syndrome, he also underwent 3.5 parathyroidectomy, as well as adenomectomy, followed by calcium replacement therapy because of postoperative hypocalcaemia. Under treatment with lanreotide and cabergoline for 2 years, the patient’s IGF-1 level was still elevated (423 ng/mL) and pituitary MRI was repeated. MRI showed two microadenomas measuring 9 mm and 7 mm. Surgery was repeated via the transsphenoidal route in 2010. The immunhistopathology report displayed that the pituitary adenoma was stained with GH, and prolactin. Ki67 index was 4%. Early postoperative MRI showed no residual adenoma. After the operation, disease control was achieved and IGF-1 level was decreased to 89 ng/mL and stayed within normal range for 2 years. The patient was subsequently lost to follow-up for 2 years.
During routine surveillance of acromegaly from 2010–2012, a liver mass at segment 8, which enhanced maximally in the early arterial phase and became isodense in the portal venous phase, was recognised on computed tomography (CT) scans. Consequently, abdominal MRI was performed, followed by an octreotide scan, and revealed two focal areas of increased tracer uptake at the wall of duodenum. The liver lesion, which was defined at both CT and MRI, showed no activity. Gastrin level was also checked, but it was within the range of normal limits. Both duodenal and liver lesions were evaluated as suggestive of a non-functioning NET and its metastases. In addition to that, upper gastrointestinal endoscopy showed multiple lesions at duodenum. Some part of tumour was resected and pathology reported the lesion as grade 1 duodenal NET.
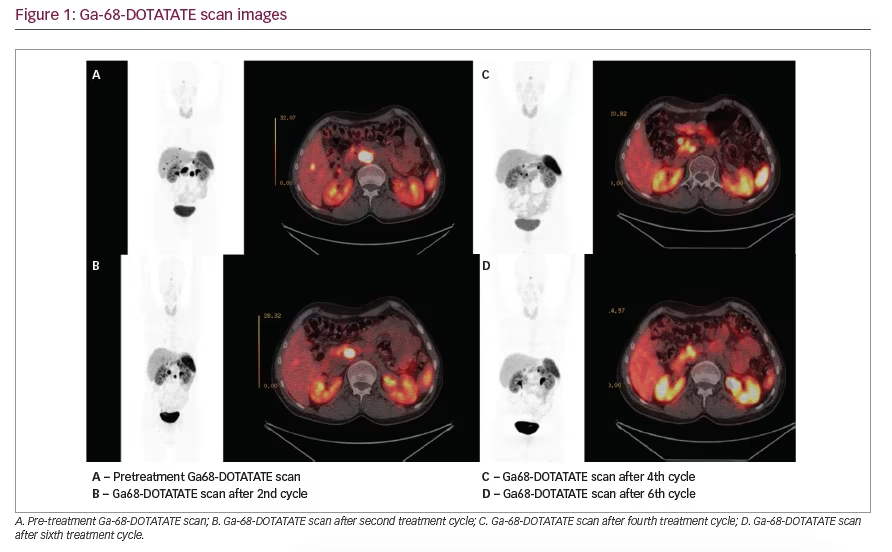
The patient was lost to follow-up for the next 3 years; however, he was referred back to our hospital in 2015. His IGF-1 level was 310 ng/mL, so lanreotide 120 mg/month was reinitiated. There also appeared liver lesions on abdominal MRI. Thereafter, a Ga-68-DOTATATE scan was performed, which revealed multiple foci with elevated accumulation of Ga-68 (Figure 1). As a result, PRRT with 177Lu-DOTATATE was administered in six cycles. Repeated Ga-68-DOTATATE scanning after each two cycles displayed an excellent response to PRRT (Figure 1); additionally, no side effect of this treatment, such as myelosuppression or radiation nephritis, was observed.
Discussion
PRRT is one of the systemic radiotherapies that allows accumulation of radionuclides in the tumour cells which express high levels of somatostatin receptors. There are two types of radiopeptides used for PRRT, 90Y and 177Lu. As the most widely used radiopeptide for PRRT,
177Lu-DOTATATE treatment is a valuable therapeutic option for metastasized or inoperable GEP-NETs which harbour the somatostatin receptors on their surface. Inclusion criteria for use of PRRT with 177Lu-DOTATATE in NETs is simply presenting an inoperable/metastatic tumour with sufficient tumour uptake on the diagnostic somatostatin receptor scintigraphy. In addition to that, the patient should have >50 mL/min creatinine clearance, >50 Karnofsky performance status, sufficient bone marrow reserves and >3 months expected survival.4
The first-line of treatment for the metastatic GEP-NET in this case may be debatable. There appeared one metastatic lesion on the liver at segment 8, for which surgery could have also been considered. Chakedis et al. examined 581 patients with metastatic NETs in various organs including the liver (61.3%), who underwent resection. They reported that removing all metastatic disease had better prognosis than debulking or palliative resection (112.5, 89.2 and 50.0 months, respectively, p<0.001).5 Despite these promising results, the multidisciplinary council at our clinic decided to use 177Lu-DOTATATE for the management of the metastatic disease. The primary reason for this consensus was the elevated accumulation of Ga-68 at described lesions and the young age of our patient. A systemic therapy, not as risky as surgery, was considered first and if it failed, the surgery would be an alternative option.
As described earlier, the NETTER-1 study is the first randomised phase III study to compare 177Lu-DOTATATE to high-dose octreotide long-acting repeatable (LAR) therapy in patients with inoperable and progressive midgut NETs.6 In this trial, 229 patients with inoperable, metastatic, well-differentiated somatostatin-receptor-positive midgut NETs who had progressive disease on standard dose (30 mg every
3–4 weeks) of octreotide LAR were included. They were randomised to an additional four cycles of 177Lu-DOTATATE (200 mCi each) every 8 weeks or octreotide LAR 60 mg alone every 28 days (control group). The estimated PFS rate at month 20 was 65.2% in the 177Lu-DOTATATE group and 10.8% in the control group. These results clearly indicated that the treatment with 177Lu-DOTATATE had a significantly higher response rate than high-dose octreotide LAR among patients with advanced midgut NETs. Another promising outcome was the limited clinically significant myelosuppression rate with 177Lu-DOTATATE treatment which was less than 10% of patients.
Furthermore, one of the largest studies which involved over 1,200 patients with advanced grade 1–2 GEP or lung NETs (with a Ki67 index of 20% or below) treated with 177Lu-DOTATATE demonstrated the excellent efficacy and limited transient side effects of this therapeutic option. There was no therapy-related long-term renal or hepatic failure.6 In another study of 265 patients with NETs reported in 2011, it was stated that the quality of life improved significantly after 177Lu-DOTATATE therapy.7
In conclusion, the dramatic response to PRRT with 177Lu-DOTATATE described in this case report, and recent published articles indicating the beneficial efficacy and limited adverse effects of 177Lu-DOTATATE, should encourage clinicians to use PRRT for inoperable or metastatic NETs. Complete blood count, renal function tests, performance status of the patient and somatostatin receptor scintigraphy images should be assessed cautiously before initiating this treatment.