Renal tubular acidosis (RTA) is characterized by the impaired ability of the kidneys to absorb filtered bicarbonate or to excrete titrable acid in urine.1 It manifests as normal anion gap (hyperchloremic) metabolic acidosis. RTA may be of four types: type 1 (distal RTA), type 2 (proximal RTA), type 3 (mixed RTA) and type 4.1 Type 1 distal RTA is caused by impaired ability of the α intercalated cells of the kidney to excrete hydrogen ions in urine.2 This leads to systemic acidosis with an alkaline urine pH. Type 2 proximal RTA is caused by impaired bicarbonate absorption, leading to excess bicarbonate excretion in urine and systemic acidosis.2 It can be either an isolated renal tubular defect or part of a generalized tubular defect known as Fanconi’s syndrome. Type 3 (both proximal and distal or mixed) RTA is rare and is usually seen in an autosomal recessive disease due to inherited carbonic anhydrase deficiency.1 Type 4 RTA is caused by hyporeninaemic hypoaldosteronism manifesting as hyperkalaemia and metabolic acidosis, which is commonly seen in diabetes.1 Type 4 RTA is generally seen in mild to moderate chronic glomerular insufficiency. Aldosterone acts on distal renal tubules to reabsorb sodium and excrete potassium. In aldosterone deficiency, the kidneys’ ability to excrete potassium is impaired. Hyperkalaemia, in turn, impairs renal ammoniagenesis, leading to impaired urinary ammonium excretion, while the excretion of protons remains in tact. Uncorrected acidosis may lead to excess osteoclastic bone resorption. Simultaneously, tubular dysfunction may also cause hypokalaemia, hypophosphataemia, vitamin D deficiency and secondary hyperparathyroidism.3,4 These may culminate in severe metabolic bone disease (MBD) in the form of rickets, osteomalacia, pathological fractures and secondary osteoporosis. Here, we present our experience with five cases of RTA whose primary manifestation was severe MBD.
Case presentation
Patient 1
An 18-year-old female presented with progressive deformity of bilateral lower limbs in the form of genu valgum from the age of 2 years (Figure 1A). She also developed progressive proximal myopathy and diffuse skeletal pain. Two months prior to presentation, she started experiencing continuous pain of severe intensity in the left thigh after suffering from a trivial trauma. She then consulted an orthopaedician and underwent radiographs that revealed multiple insufficiency fractures (Figure 1B). She has been bedridden for the past 2 months.
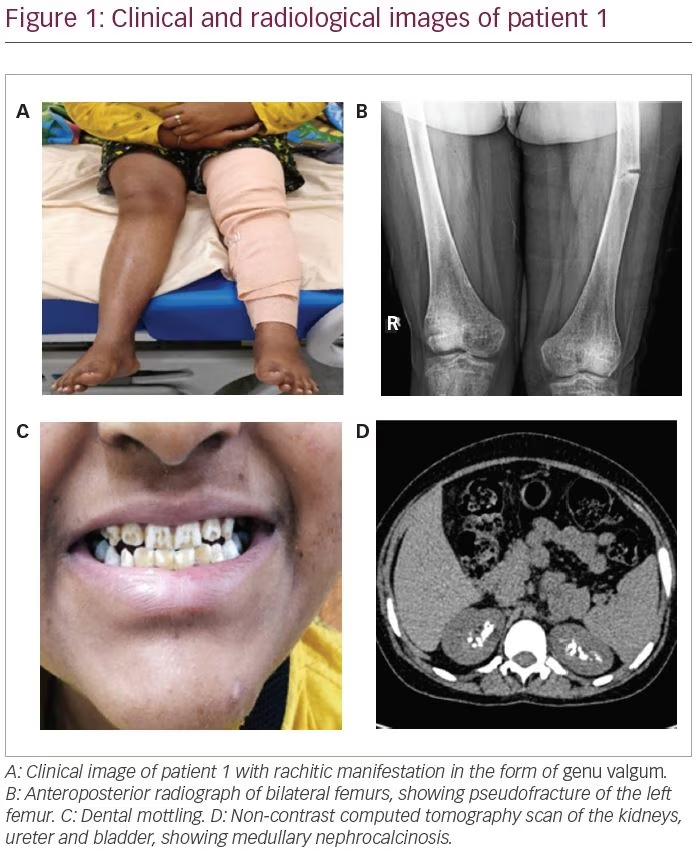
The patient also gave history of polyuria and nocturia and craving for savoury foods. She was the shortest among all her family members and peer groups. She had taken intermittent courses of oral calcium and vitamin D supplements for the past 6 years. She had normal pubertal development with a Tanner stage of B5 and P5. She had dental mottling and dental attrition (Figure 1C), although there was no history of dental abscess or premature tooth loss. None of her family members had a similar illness.
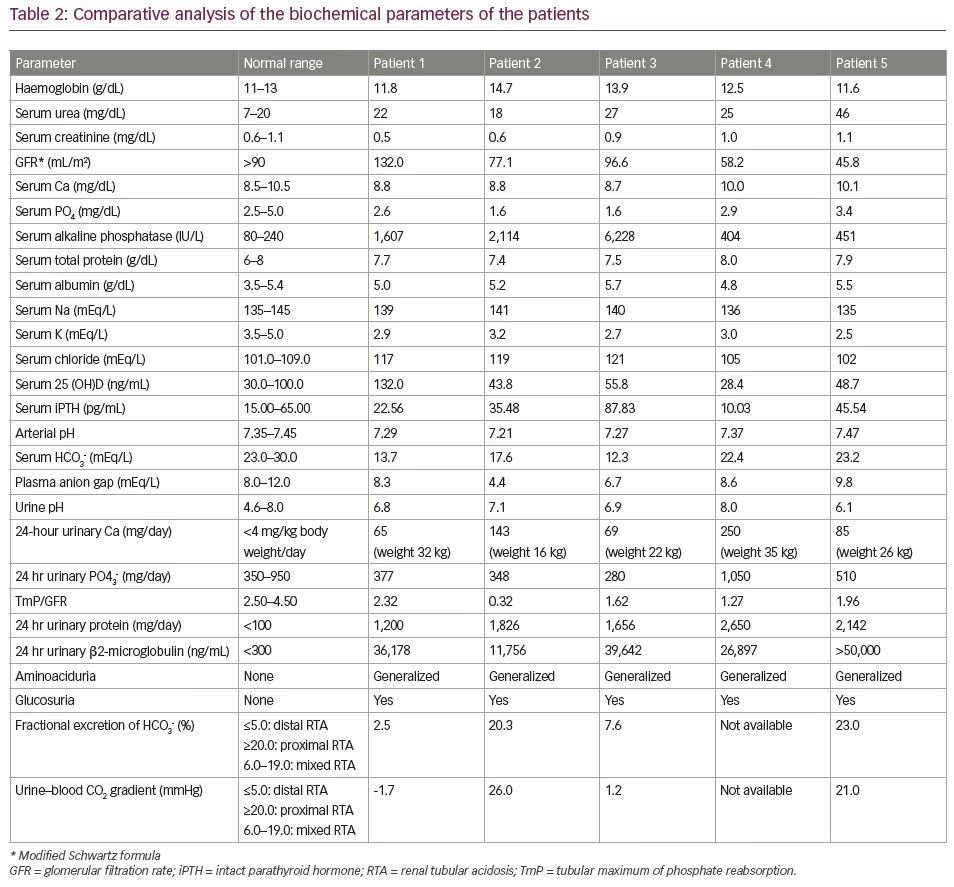
Biochemically, she had hypokalaemia (2.9 mEq/L; normal range [N]: 3.5–5.0 mEq/L) and low normal serum phosphate (2.6 mg/dL; N: 2.5–5.0 mg/dL) with low tubular maximum of phosphate reabsorption (TmP) for glomerular filtration rate (GFR) (TmP/GFR: 2.3; N: 2.5–4.5) indicating renal phosphate loss. She had normal total calcium (8.8 mg/dL; N: 8.5–10.5 mg/dL), elevated 25-hydroxy vitamin D (25 (OH)D) (132 ng/mL; N: 30–100 ng/mL) and normal intact parathyroid hormone (iPTH) levels (22.5 pg/mL; N: 15–65 pg/mL). Her serum alkaline phosphatase was markedly elevated (1,607 IU/L; N: 80–240 IU/L), which is suggestive of active MBD. Her serum chloride levels were elevated (117 mEq/L; N: 101–109 mEq/L).
Further workup revealed features of distal and proximal RTA. Her urine pH was 6.8 (N: 4.5–7.8) with an arterial pH of 7.29 (N: 7.35–7.45) with normal plasma anion gap of 8.1 mEq/L (N: 8–12 mEq/L). In the presence of normal anion gap systemic metabolic acidosis, the patient had alkaline urine that suggested distal renal tubular involvement. Simultaneously, she also had features of proximal RTA with serum bicarbonate of 13 mEq/L (N: 23–30 mEq/L), along with low molecular weight proteinuria (urinary β2-microglobulin 36,178 ng/mL; N: <300 ng/mL), glucosuria and generalized aminoaciduria (presence of all amino acids in urine). To differentiate between proximal and distal RTA, the patient underwent a bicarbonate loading test. At a loaded state of urine pH >7.5 and serum HCO3 >20 mEq/L, fractional excretion of bicarbonate (FeHCO3-) was calculated, which was 2.5%, and the urine–blood CO2 gradient was calculated, which was -1.7 mmHg; both suggested predominant distal tubular involvement. Although she did not have hypercalciuria, ultrasonography and non-contrast computed tomography of the kidney revealed bilateral medullary nephrocalcinosis (Figure 1D) – another hallmark of distal RTA.
Thus, a diagnosis of MBD secondary to RTA was considered. The patient had features of mixed RTA (type 3), although the FeHCO3- and urine–blood CO2 gradient suggested predominant distal involvement.
In this patient, there was no history of exposure to any drug or toxin, and ophthalmological examination did not reveal any lenticular opacity (galactosaemia, Lowe’s syndrome) or Kayser–Fleischer ring (Wilson’s disease).5 Tests for systemic lupus erythematosus ([SLE] anti-double stranded [ds]DNA), rheumatoid arthritis (rheumatoid arthritis factor, anti-cyclic citrullinated peptide), chronic active hepatitis and primary biliary cirrhosis were negative. Primary Sjögren’s syndrome was also ruled out with normal autoimmune markers (normal anti-Sjögren’s-syndrome-related antigen A [SSA], anti-Sjögren’s-syndrome-related antigen B autoantibodies [SSB], normal Schirmer test). Human immunodeficiency virus (HIV), hepatitis B, hepatitis C were also ruled out with normal serological tests.5 Serological analysis of heavy metals could not be done due to resource constraints. However, none of the family members or anyone in the neighbourhood who consumed water from the same source had similar disease.
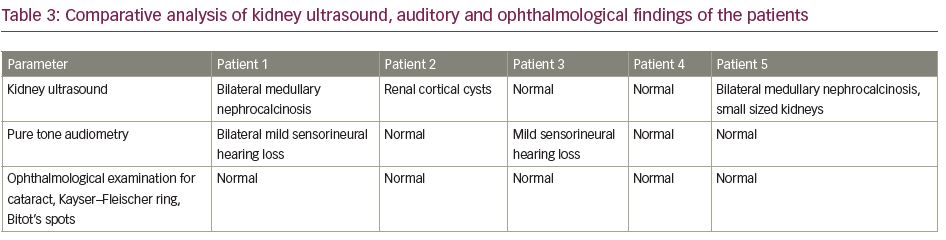
Early onset of disease manifestation suggested a possibility of genetic aetiology. Mutations of genes such as SLC4A4, SLC4A1, ATP6V1B1 and ATP6V0A4, which encode for renal tubular channel proteins, may give rise to various inherited forms of RTA.5 However, the family was unwilling to undergo genetic analysis due to financial constraints. Although the patient did not complain of any hearing impairment, pure tone audiometry revealed bilateral mild sensorineural hearing loss, which again is more common in genetic variants of RTA.6
The patient was started on potassium citrate solution with bicarbonate content of 3 mEq/kg body weight/day and phosphate supplement of 30 mg/kg/day. Phosphate solution was prepared by mixing of disodium hydrogen phosphate and sodium dihydrogen phosphate powder in water. It was administered in three divided doses, and freshly prepared before each dose. A low dose of active vitamin D was also added (calcitriol 0.25 µg/day). After the initiation of treatment, the patient developed diarrhoea following which the phosphate dose was reduced. She was asked to follow-up after an interval of 3 months as the plan was to monitor for clinical, biochemical and radiological improvement.
Patient 2
A 14-year-old female presented with complaints of progressive windswept deformity of lower limbs from the age of 3 years (Figure 2A). She also had diffuse skeletal pain, proximal myopathy and significant short stature. There was no family history of similar disease.
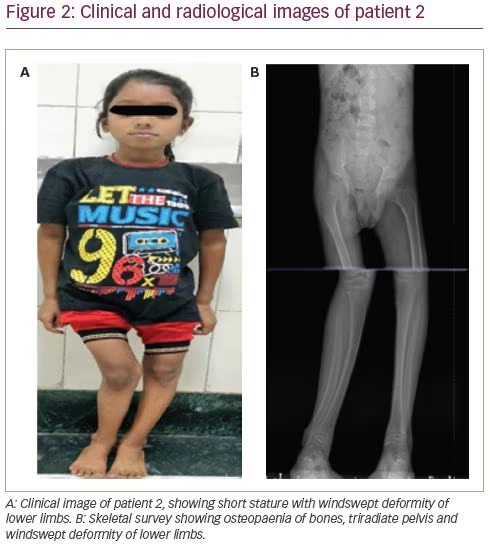
On examination, she had disproportionate short stature with a corrected height of 112 cm and weight of 16 kg. Biochemically, she had normal total calcium (8.8 mg/dL), severe hypophosphataemia (1.6 mg/dL; N: 2.5–5.0 mg/dL) with low TmP/GFR (0.3), low potassium (3.2 mEq/L; N: 3.5–5.0 meq/L) and markedly elevated alkaline phosphatase (2,114 IU/L). She had normal 25(OH)D levels (43.8 ng/mL; N: 30–100 ng/mL) and normal iPTH (35.4 pg/mL). Similarly to the first patient, she had features of both proximal and distal RTA. Baseline urine pH was 6.3 with an arterial pH of 7.21. Serum bicarbonate was 17.6 mEq/L with plasma anion gap of 4.4 mEq/L and elevated serum chloride levels of 119 mEq/L. She also had generalized aminoaciduria, glucosuria and low molecular weight proteinuria (urinary β2-microglobulin: 11,756 ng/mL). A bicarbonate loading test was done to differentiate between proximal and distal RTA. At a loaded state, her FeHCO3- was 20.3% and the urine–blood CO2 gradient was 26 mmHg, both suggesting predominant proximal involvement. Although the patient had hypercalciuria, there was no nephrocalcinosis. There was also no hearing loss evident on pure tone audiometry. A skeletal survey revealed a triradiate pelvis with pseudofractures and windswept deformity of the lower limbs (Figure 2b).
Aetiologies of RTA, such as autoimmune diseases (Sjögren’s, SLE, rheumatoid arthritis), were ruled out with normal autoimmune markers (normal anti-nuclear antibodies, anti-dsDNA, anti-SSA, anti-SSB), and ophthalmological examination did not reveal any lenticular opacity (galactosaemia, Lowe syndrome) or Kayser–Fleischer ring (Wilson disease). Serological tests for HIV, hepatitis B and hepatitis C were negative. Liver function tests were normal with no evidence of primary biliary cirrhosis or chronic active hepatitis. Renal function tests were also normal with no evidence of obstructive or reflux nephropathy. There was no history of chronic exposure to any drug (amphotericin B, lithium, analgesic abuse, toluene, amiloride, trimethoprim, pentamidine, vanadium) or heavy metal (lead, cadmium, mercury) known to cause RTA. A biochemical analysis of heavy metals was not performed due to lack of evidence of the clustering of disease in the family or neighbourhood. The possibility of genetic aetiology was not discounted owing to early onset of the disease. However, due to resource constraints, genetic analysis could not be done.
This patient was started on potassium citrate to provide a bicarbonate concentration of 10 mEq/kg/day, along with a phosphate supplement of 30 mg/kg/day in divided doses. The bicarbonate dose administered was higher compared to that in the first patient, due to predominant proximal tubular involvement (significant bicarbonaturia). Calcitriol was not added as she had hypercalciuria. With this treatment, she was discharged and asked to follow-up after an interval of 3 months.
Patient 3
A 21-year-old male was reportedly physically fit until the age of 16 years, when he developed diffuse skeletal pain of moderate to severe intensity. This was followed by features of proximal myopathy and progressive windswept deformity of the lower limbs. Subsequently, he was unable to bear weight on his lower limbs. During this time, the patient ambulated by squatting and putting pressure on his upper limbs. He then developed outward deformity of bilateral upper limbs and prominence of his chest wall, shoulder blades and clavicles (Figure 3A). He also lost significant height and weight over the course of 5 years. Similarly to the first patient, he too had polyuria, nocturia and a craving for savoury foods. He also complained of intermittent night blindness. He was bedridden for 3 years prior to presentation to the clinic at the age of 21 years, and was unable to carry out daily activities without help. Although he was significantly emaciated and had severe musculoskeletal symptoms, he did not seek treatment from a physician; this led to a delayed diagnosis of bilateral femur neck fractures.
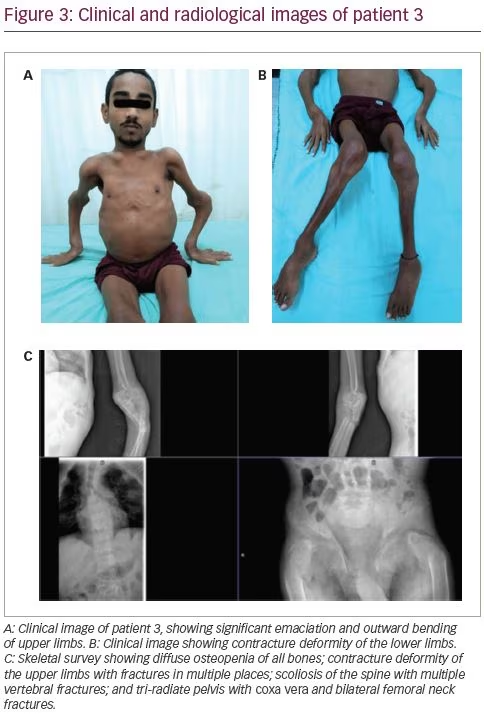
On examination, he was emaciated. He had pectus carinatum, rachitic rosary, prominence of clavicles and shoulder blades, contracture deformity of the upper limbs and severe proximal myopathy with a power of 3/5 in all proximal muscles. He had permanent flexion contracture at the knees with windswept deformity (Figure 3B). There was tenderness at all the bony prominences. His treatment history included multiple intermittent courses of calcium and vitamin D supplements.
Biochemical workup revealed normal total calcium (8.7 mg/dL) with hypokalaemia (2.7 mEq/L) and hypophosphataemia (1.6 mg/dL) with low TmP/GFR (1.6), indicating renal phosphate loss and immensely elevated serum alkaline phosphatase (6,228 IU/L). He had normal 25(OH)D levels (55.8 ng/mL) with a mildly elevated intact PTH level (87.8 pg/mL). A skeletal survey revealed multiple rib fractures, multiple pseudofractures in the upper limbs, bilateral femoral neck fractures, coxa vera and windswept deformity of the lower limbs (Figure 3C).
Workup revealed features of both distal RTA (urine pH 6.9 in the presence of normal anion gap systemic acidosis; i.e. blood pH: 7.27; plasma anion gap: 6.7 mEq/L) and proximal RTA (hypophosphataemia with low TmP/GFR [-1.62], low molecular weight proteinuria [urinary β2-microglobulin: 39,642 ng/mL], glucosuria and generalized aminoaciduria). A bicarbonate loading test suggested redominant distal renal tubular involvement (FeHCO3-: 7.6%; urine–blood CO2 gradient: 1.2 mmHg). There was no hypercalciuria or nephrocalcinosis. An aetiological workup ruled out autoimmune diseases (negative autoimmune markers for SLE, rheumatoid arthritis, Sjögren’s syndrome), HIV, and hepatitis B and C infection. Antibodies for Sjögren’s syndrome (anti-SSA and anti-SSB) and Schirmer’s test were normal. There was no ophthalmological evidence of galactosaemia, Lowe’s syndrome or Wilson’s disease. There was also no evidence of chronic active hepatitis, primary biliary cirrhosis or obstructive uropathy or reflux nephropathy. There was also no prolonged exposure to any offending drug or heavy metal. Unlike the previous two patients, who had an early onset of disease, this patient had an onset of manifestations in late adolescence. Certain inherited forms of RTA – like autosomal dominant mutations of genes encoding the distal tubular channels, such as ATP6V1B1 or ATP6V0A4 – may be associated with late onset of disease.7 However, again due to a resource-poor setting, a genetic analysis was not done.
This patient was also started on phosphate (30 mg/kg/day) in divided doses and potassium citrate (bicarbonate content 3 mEq/kg/day) supplements along with low-dose calcitriol (0.25 µg/day). He developed diarrhoea after the initiation of phosphate; subsequently, his dose was reduced.
Patient 4
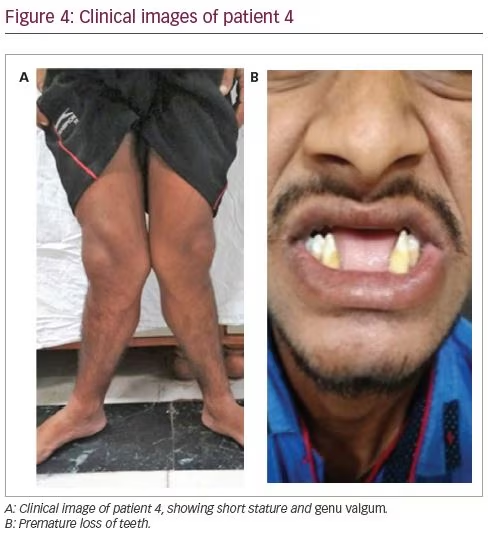
A 23-year-old male had been symptomatic since the age of 4 years. He had progressive genu valgum and significant short stature (Figure 4A). This was accompanied by diffuse musculoskeletal pain and proximal myopathy. He had recurrent dental abscesses and premature loss of teeth (Figure 4B). Similarly to patient three, he had intermittent night blindness. There was no episode of hypokalaemic periodic paralysis. A biochemical workup revealed features of incomplete distal RTA (urine pH: 8.0; blood pH: 7.37). He also had hypokalaemia (3.0 mEq/L) and hypercalciuria (urinary Ca >4 mg/kg/day), but no nephrocalcinosis or renal stones. There was no systemic acidosis, but on a furosemide–fludrocortisone test, his urine pH remained >5.3 even after 5 hours. For further confirmation of acidosis, an ammonium chloride (NH4Cl) test was done. Acidosis was induced with NH4Cl 100 mg/kg dose mixed in fruit syrup in breakfast, which was consumed over 2 hours. After 6 hours of administration, his blood pH fell to <7.3, while his urine failed to acidify (urine pH >5.3). This confirmed the presence of incomplete distal RTA. He also had features of proximal RTA in the form of low normal serum phosphate (2.9 mg/dL) with low TmP/GFR (1.27), low molecular weight proteinuria urinary β2-microglobulin: 26,897 ng/mL), glucosuria and generalized aminoaciduria.
An aetiological workup did not reveal any specific cause of RTA. Considering the late onset of the disease, it was important to rule out acquired causes, which were sequentially evaluated. Tests for autoimmune diseases (anti-nuclear antibodies, anti-dsDNA, anti-SSA, anti-SSB) and Schirmer’s test for Sjögren’s syndrome were negative. Genetic analysis was not done due to resource constraints.
The patient was managed with potassium citrate solution with a bicarbonate content of 3 mEq/kg/day and a phosphate supplement of 30 mg/kg/day in three divided doses. Calcitriol was not added owing to concurrent hypercalciuria.
Patient 5
A 14-year-old male had progressive deformity of the lower limbs in the form of genu valgum from the age of 9 years. He was also the shortest among all his peers. There was a history of polyuria and nocturia. There was no history suggestive of proximal myopathy, fracture, hypokalaemic periodic paresis, renal calculi or graveluria.
On examination, he had significant disproportionate short stature with a height of 122 cm, and a weight of 26 kg. He had genu valgum with an intermalleolar distance of 10 cm. A biochemical workup revealed a normal blood pH of 7.47 with a urine pH of 6.1. Acidosis was induced with furosemide 40 mg and fludrocortisone 1 mg. At the end of 5 hours, the urine pH remained at >5.3, suggesting incomplete distal RTA. This was followed by the confirmatory NH4Cl test with the urine failing to acidify (urine pH >5.3) after 6 hours of NH4Cl administration. He also had other features of distal RTA, such as hypokalaemia and hypercalciuria. Ultrasonography revealed bilateral small-sized kidneys with medullary nephrocalcinosis. In addition, he had proximal tubular involvement in the form of low molecular weight proteinuria (urinary β2-microglobulin >50,000 ng/mL) and generalized aminoaciduria. The patient had normal serum phosphate levels (3.4 mg/dL). Bicarbonate loading test revealed predominant proximal tubular involvement with FeHCO3- of 23% and a urine–blood CO2 gradient of 21 mmHg.
An aetiological workup ruled out autoimmune diseases, such as SLE, rheumatoid arthritis, Sjögren’s syndrome; viral infections, such as HIV and hepatitis B and C; and genetic diseases such as Wilson’s disease, galactosaemia and Lowe’s syndrome. There was no history of any liver or renal disease. There was also no history of prolonged exposure to any drug or heavy metal. Genetic analysis for autosomal dominant or recessive RTA could not be performed due to resource constraints. The patient was started on potassium citrate with a bicarbonate content of 3 mEq/kg/day. Phosphate was not added as the serum phosphate level was normal. Calcitriol was also not added due to concomitant hypercalciuria.
Discussion
We have described five patients who presented to us with severe MBD at various age groups. The clinical features are summarized in Table 1 and the biochemical features of the patients are mentioned in Table 2. Ophthalmological, auditory and ultrasound kidney findings are summarized in Table 3. While evaluating for the aetiology of MBD, all were found to have RTA. All of them had features of both proximal and distal RTA (mixed type or type 3). True mixed RTA is rare and is found in the autosomal recessive disease due to carbonic anhydrase deficiency (CA2 gene mutation). However, proximal tubular dysfunction may occur secondary to distal RTA. This is usually a transient phenomenon, which does not represent a distinct genetic entity, and is usually related to some exogenous factor, such as high salt intake.8 On follow-up, there is usually recovery of proximal tubular dysfunction after treatment with alkali therapy. Only very few case reports depicting the pattern of mixed RTA exist.9,10
The clinical features to suggest RTA include growth retardation, failure to thrive, polyuria, polydipsia, preference for savoury foods, refractory rickets, renal calculi, nephrocalcinosis and unexplained hypertension.11
RTA should always be considered in the differential diagnosis of rickets non-healing to calcium and vitamin D supplements. Osteoclasts require an acidic environment between their ruffled border and bone mineral surface for initiation of resorption. In the presence of uncorrected systemic acidosis, there is increased RANKL expression promoting the differentiation of pre-osteoclasts to osteoclasts with the stimulation of osteoclastic bone resorption uncoupled with bone formation.4 In proximal tubular involvement, there is also impaired phosphate handling causing hypophosphataemia. Hypophosphataemia impairs mineralization of the cartilage plate, leading to expansion of the growth plate. Bone deformities develop when excessive weight is induced on the unmineralized growth plates. Hence, these deformities are usually noticed when the child starts crawling or walking. Acidosis also impairs vitamin D metabolism and action leading to hypophosphataemia and hypocalcaemia, resulting in impaired mineralization of the epiphyseal growth plate.
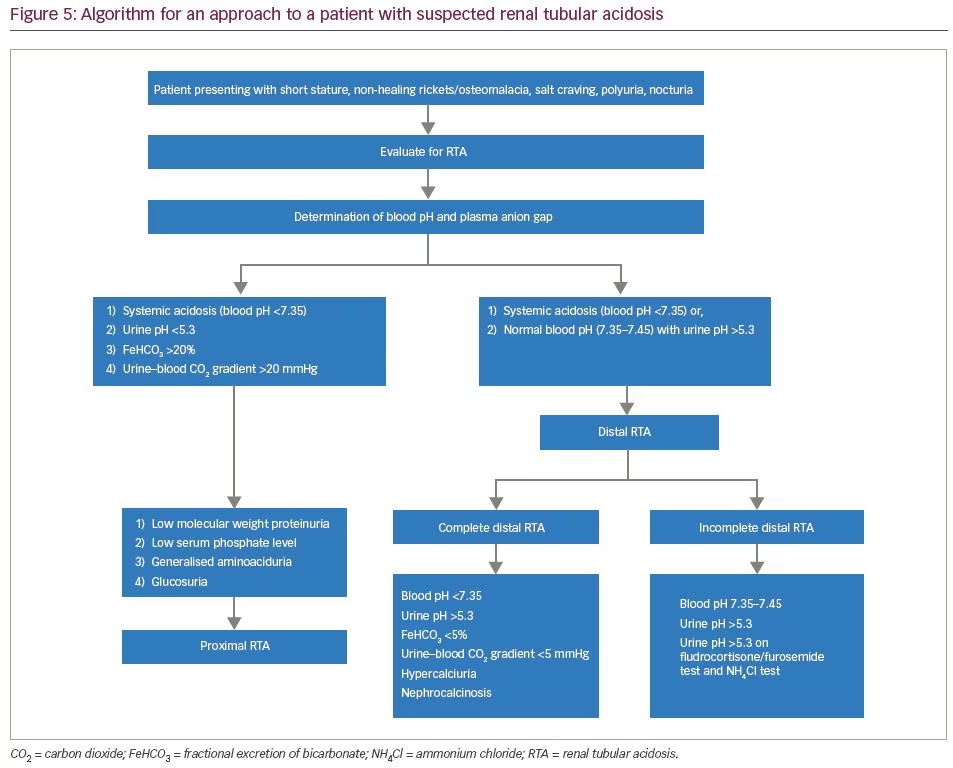
For evaluation of RTA, the first step is the determination of the plasma anion gap. RTA causes a normal anion gap (8–12 mEq/L) hyperchloremic metabolic acidosis, which was observed in all our patients. The next step involves the determination of urinary anion gap (urinary anion gap = UNa + UK-UCl). This is followed by the determination of fasting urinary pH. In the presence of systemic metabolic acidosis, urinary pH more than 5.3 suggests defective H+ secretion, indicating distal RTA.11 In incomplete distal RTA, patients may not have systemic acidosis. In such circumstances, it is useful to proceed with the furosemide/fludrocortisone test and the NH4Cl test. Furosemide/fludrocortisone is a good ‘rule out’ test to exclude distal RTA.12 After an overnight fast, morning urinary pH is calculated. Subsequently, the patient is treated with 40 mg of furosemide and 1 mg of fludrocortisone. The principle of this test lies in the fact that furosemide blocks the sodium channel transporter NKC22 in the thick ascending limb of the loop of Henle.13 Subsequently, there is a loading of sodium in the distal collecting tubules and collecting duct. The addition of fludrocortisone enhances sodium reabsorption in collecting duct epithelial cells, and directly enhances the activity of H+-ATPase. This leads to the excretion of proton ions in urine leading to urinary acidification. However, patients with incomplete distal RTA lack the ability of urinary acidification, hence urine pH remains >5.3 after 5–6 hours of furosemide/fludrocortisone administration.
Ammonium chloride (NH4Cl) loading is the confirmatory test for incomplete distal RTA.14 Acidosis can be induced by the administration of a certain quantity of NH4Cl that produces a decline in blood pH and blood CO2 concentration. In normal individuals, with systemic acidosis, there is excretion of H+ ions in urine, leading to urinary acidification. However, in patients with incomplete distal RTA, there is impaired secretion of protons in urine, and urine pH remains >5.3. The NH4Cl test is cumbersome to use owing to gastric irritation, nausea and unpalatability. An algorithm for the approach to a suspected case of RTA is shown in Figure 5.
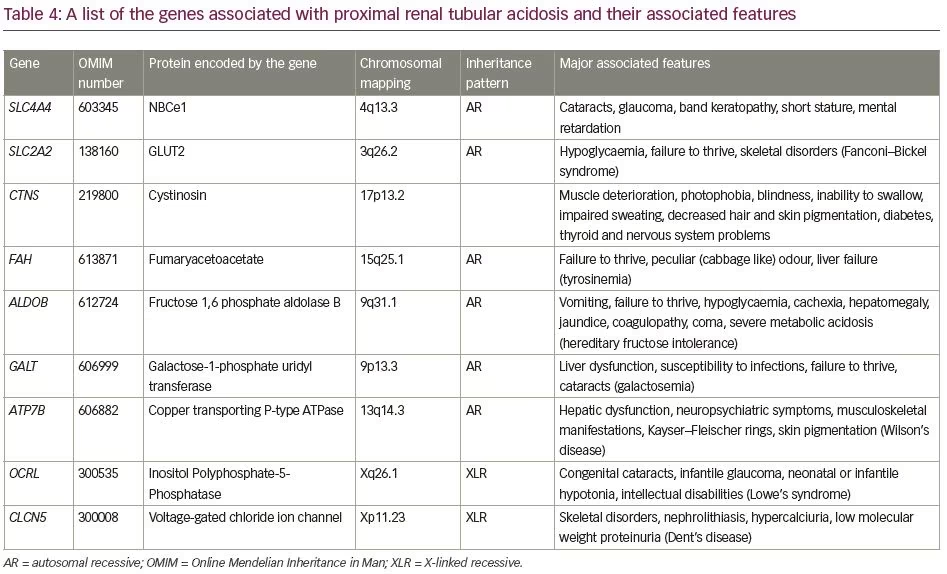
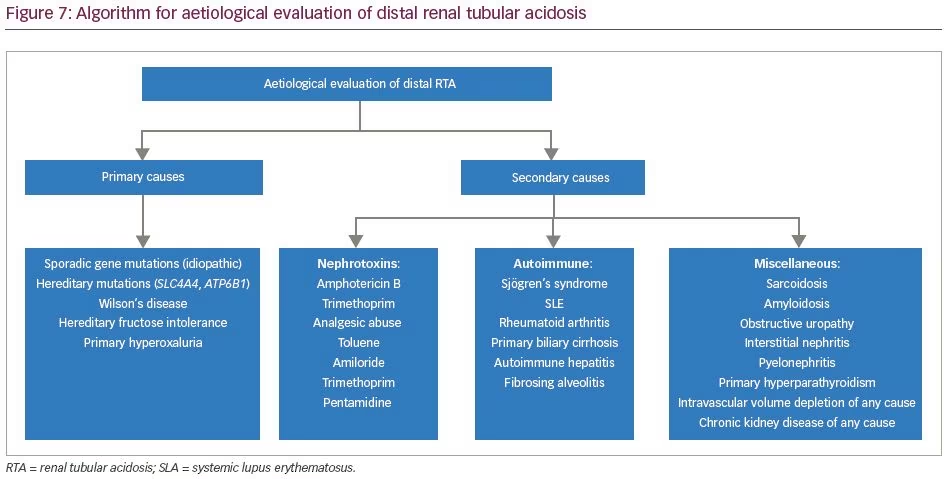
The aetiologies of proximal RTA include primary causes, such as mutations of genes SLC4A4 and SLC2A2; secondary causes in the context of Fanconi’s syndrome (e.g. cystinosis, galactosaemia, fructose intolerance, Wilson’s disease, Lowe’s syndrome, multiple myeloma and light chain disease); drugs and toxins (e.g. acetazolamide, outdated tetracycline, aminoglycoside, valproate, 6-mercaptopurine, streptozotocin, ifosfamide, lead, cadmium and mercury); or other clinical entities, such as vitamin D deficiency, hyperparathyroidism, medullary cystic disease, Alport’s syndrome, cortico-resistant nephrotic syndrome, renal transplantation, amyloidosis and recurrent nephrolithiasis.5 Aetiologies of distal RTA include primary causes, such as autosomal dominant or recessive mutations of genes SLC4A1, ATP6V1B1, ATP6V0A4; secondary causes in the context of genetic diseases (e.g. Wilson’s disease, hereditary fructose intoleranc and primary hyperoxaluria), calcium disorders (e.g. primary hyperparathyroidism, vitamin D intoxication and idiopathic hypercalciuria with nephrocalcinosis), autoimmune diseases (e.g. SLE, Sjögren’s syndrome, chronic active hepatitis, primary biliary cirrhosis, thyroiditis, fibrosing alveolitis and rheumatoid arthritis), renal diseases (e.g. renal transplant rejection, medullary sponge kidney, obstructive and reflux nephropathy, and Balkan nephropathy); and drugs and toxins (e.g. amphotericin B, lithium, analgesic abuse, toluene, amiloride, trimethoprim, pentamidine and vanadium).5 In our patients, all common causes of RTA were ruled out. Genetic aetiologies were suspected, but due to resource constraints, genetic testing could not be performed in our patients. A comprehensive list of the genes associated with proximal and distal RTA and their associated manifestations is shown in Table 4 and Table 5, respectively.
An algorithm for the evaluation of proximal RTA is shown in Figure 6. An algorithm for evaluation of distal RTA is shown in Figure 7. All our patients had features of combined proximal and distal RTA. Tests that differentiate between the two are the bicarbonate loading test and the urine–blood CO2 gradient in a bicarbonate-loaded state.11
Bicarbonate loading is administered with either intravenous or oral bicarbonate. In a bicarbonate-loaded state (i.e. urine pH >7.5 or serum bicarbonate >20 mEq/L) the fractional excretion of bicarbonate should be calculated [FeHCO3 = (urine HCO3/serum HCO3) × (serum creatinine/urine creatinine)]. FeHCO3 >20% suggests proximal RTA, <5% suggests distal RTA, and 5–20% denotes mixed RTA. Similarly, urine–blood CO2 gradient >20 mmHg suggests proximal RTA, and <20 mmHg suggests distal RTA.
The management of RTA includes correction of metabolic acidosis with bicarbonate supplements.5 Bicarbonate requirement is higher in patients with proximal RTA (5–20 mEq/kg/day) compared with distal RTA (2–3 mEq/kg/day). Alkali therapy is usually combined with potassium replacement (as potassium citrate) to avoid severe hypokalaemia.
Patients with proximal RTA often require supplements of phosphate (Joulies solution, neutral phosphate solution). Supplementation of active vitamin D in RTA has been a matter of debate. In contrast to hypophosphatemic rickets where fibroblast growth factor 23 (FGF23) levels are elevated leading to the impairment of 1-alpha hydroxylase enzyme and the generation of calcitriol, in RTA the FGF23 levels are normal. Nevertheless, acidosis itself is known to impair vitamin D metabolism and action.15-17
In our case series, patient 1 and patient 3 were treated with low-dose calcitriol. The other three patients had hypercalciuria at presentation; hence calcitriol was not initiated. Small doses of active vitamin D are required for the healing of rickets.18 Moreover, phosphate has an avidity for binding with calcium leading to hypocalcaemia and chronic parathyroid hormone stimulation. Active vitamin D also helps in the prevention of secondary and tertiary hyperparathyroidism following phosphate supplementation.
Specific therapy for an underlying disorders (e.g. cysteamine for cystinosis, D-penicillamine for Wilson’s disease, and a lactose-free diet in galactosaemia) is possible for some patients. Dietary sodium should be restricted in patients with RTA.
The follow-up of patients with RTA includes regular assessment for growth and blood levels of electrolytes, blood pH and bicarbonate. Bicarbonate levels should be kept around 20–22 mEq/L. Subjects with distal RTA are at risk for nephrocalcinosis which should be screened with ultrasound of the kidneys and 24-hour urinary calcium. Rickets should be assessed clinically, radiologically and biochemically, with a decline in serum alkaline phosphatase levels.
Conclusion
Our case series includes five patients who presented with severe MBD and were diagnosed with RTA. All of them had features of both proximal and distal RTA (type 3 [mixed] RTA). This pattern is extremely rare and is usually seen in the setting of carbonic anhydrase deficiency. However, in these patients, it might represent a transient phenomenon and not a distinct genetic entity that might be evident on follow-up. A bicarbonate loading test should be performed to differentiate between proximal and distal variants. Treatment includes the correction of acidosis with alkali supplementation along with potassium and phosphate. Low doses of active vitamin D may be supplemented in those who are not receiving hypercalciuria to help in the healing of rickets or osteomalacia, and to prevent the development of secondary/tertiary hyperparathyroidism.