Acromegaly is a chronic, progressive disease characterized by an excess secretion of growth hormone, and consequently, increased circulating insulin-like growth factor 1 (IGF-1) levels. These patients typically exhibit acral and soft tissue overgrowth, headache, arthritis and visual disturbances. Impaired glucose tolerance and diabetes mellitus are also frequently associated with acromegaly.1 We describe a patient who presented to the emergency department with diabetic ketoacidosis, and was subsequently diagnosed with acromegaly due to somatotropinoma.
Case report
A 36-year-old female presented to the emergency department with complaints of diffuse abdominal pain, vomiting and breathlessness for 2 days. She had been losing weight for 2 months, which was undocumented. On examination, she was dehydrated, her pulse rate was 110/minute, and blood pressure 100/70 mmHg. Her body weight was 60 kg and body mass index 26.9 kg/m2. The patient exhibited a Kussmaul breathing pattern with a fruity odour in her breath. Her random blood glucose was 360 mg/dL. Serum β-hydroxybutyrate levels were raised, 3.6 mmol/L (<0.6 mmol/L). Arterial blood gas analyses revealed metabolic acidosis (bicarbonate level was 8 meq/L). She was diagnosed with diabetic ketoacidosis and was treated accordingly. She responded to intravenous fluid supplementation and insulin infusion. Subsequently, treatment was changed to subcutaneous insulin therapy.
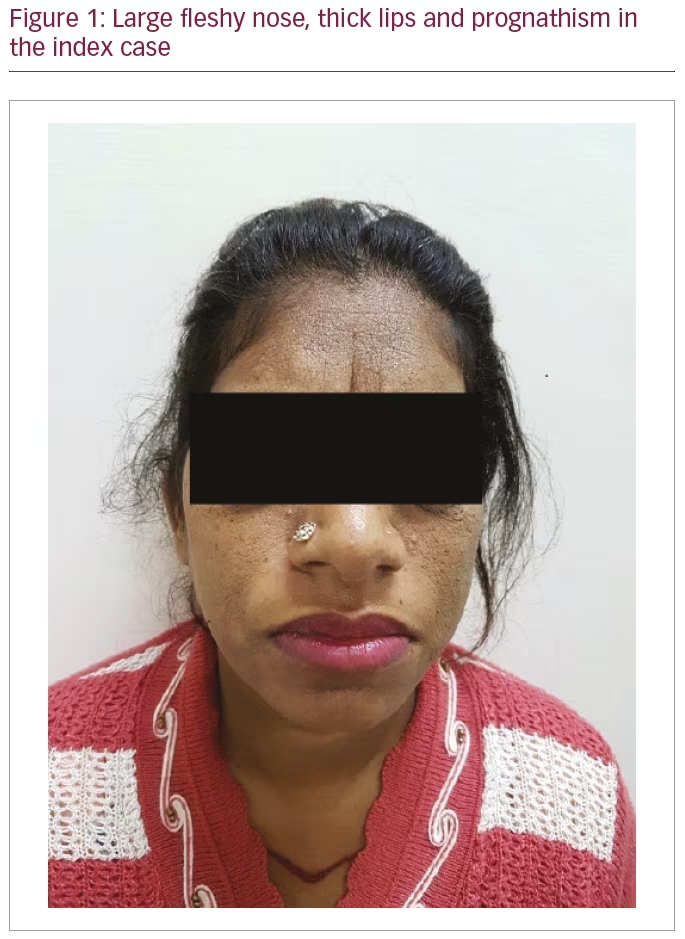
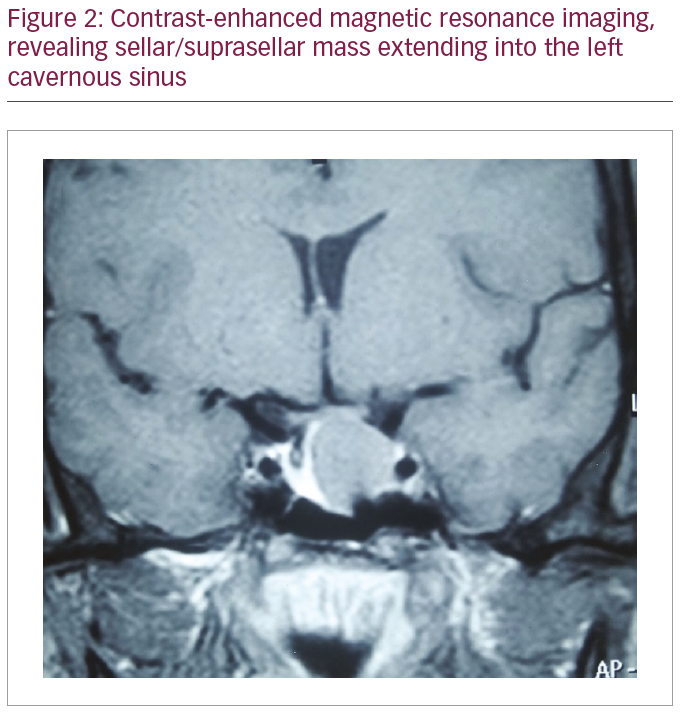
During a detailed clinical examination on initial presentation, she had coarse facial features in the form of a large fleshy nose, thick lips, prognathism and thickening of hands and feet, suggestive of acromegaly (Figure 1). Markers of insulin resistance in the form of acanthosis nigricans and skin tags were present. Investigations revealed high IGF-1 levels of 1,402 ng/mL (62–243 ng/mL) and growth hormone nadir (1 hour after a 75-g oral glucose load) was 22 ng/mL (<1 ng/mL). Complete pituitary hormonal profile revealed prolactin 22 ng/mL (4–25 ng/mL), FT3 2.8 pg/mL (2.3–4.2 pg/mL), FT4 1.2 ng/mL (0.8–1.7 ng/mL), thyroid-stimulating hormone 1.6 mU/L (0.3–5.5 mU/L), follicle-stimulating hormone 4.3 IU/L (2.5–10.2 IU/L), luteinizing hormone 6.3 IU/L (1.9–12.5 IU/L) and cortisol 218 nmol/L (100–550 nmol/L). Antibodies to glutamic acid decarboxylase were not present. Contrast-enhanced magnetic resonance imaging (MRI) of the sella revealed 20 x 18 x 15 mm sellar suprasellar mass extending into the left cavernous sinus (Figure 2).
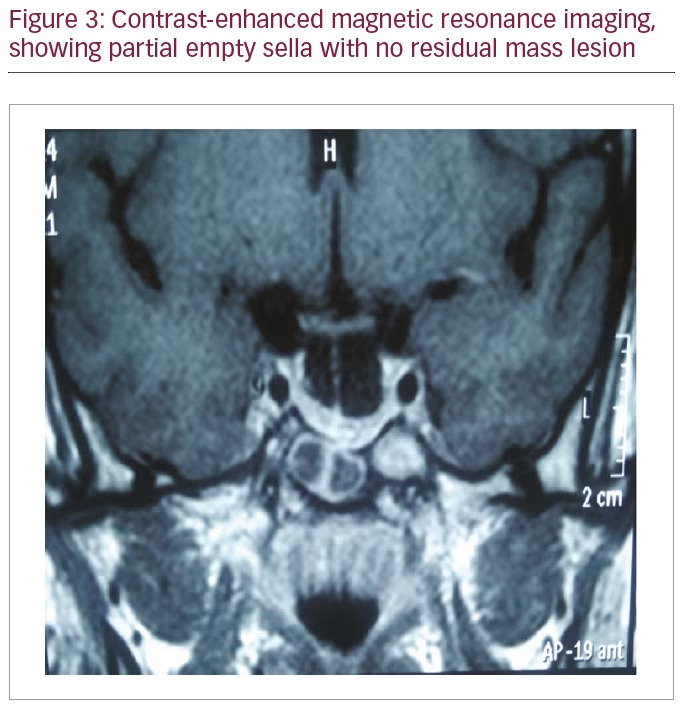
The patient underwent transsphenoidal excision of the tumour. Histopathology revealed large, round, eosinophilic cells with spherical nuclei and prominent nucleoli, suggestive of pituitary adenoma. Her blood glucose profile was normal in the immediate post-operative period without insulin. Three months post-operatively, she had normal growth hormone and IGF-1 levels. Contrast-enhanced MRI of the sella at 3 months showed partial empty sella with no definite residual/recurrent differential enhancing mass lesion (Figure 3). Her glycated haemoglobin (HbA1c) was 5.8% without insulin and oral hypoglycaemic drugs.
Discussion
In 1886, Pierre Marie published the first clinical description of disordered somatic growth and proportion, and proposed the term ‘acromegaly’.2 It is a chronic, progressive disease characterized by an excess secretion of growth hormone, and consequently, increased circulating IGF-1, typically due to a benign growth hormone-secreting pituitary adenoma.3 Patients exhibit characteristic acral and soft tissue overgrowth, headache, arthritis, respiratory obstruction, hypertension and visual disturbances from tumour mass effects. Biochemical diagnosis of acromegaly is attained by documenting non-suppressible growth hormone levels during a 2-hour period after a 75-g oral glucose load, and assessing the peripheral biologic effect of growth hormone hypersecretion by demonstrating high IGF-1 levels.1
Impaired glucose tolerance and diabetes mellitus are frequently associated with acromegaly. The prevalence of impaired glucose tolerance or impaired fasting glucose ranges from 6% to 45% and from
7% to 22%, respectively, while diabetes mellitus has been reported in 16–56% of patients with acromegaly.4,5 Both growth hormone and IGF-1 play a role in intermediate metabolism, affecting glucose homeostasis. Persistent growth hormone and IGF-1 excess can induce insulin resistance directly in the liver, adipose tissue and muscle, leading to increased gluconeogenesis, reduced glucose uptake in adipose tissue and muscle, and elevated blood glucose levels.6,7 Development of diabetic ketoacidosis requires absolute deficiency of insulin together with increased levels of counterregulatory hormones. The occurrence of diabetic ketoacidosis in acromegaly is possibly due to glucotoxicity-induced severe β-cell dysfunction, resulting in insulin deficiency. Further, high growth hormone levels have also been postulated to stimulate ketogenesis by increasing lipolysis and inhibiting fatty acid metabolism.8
There are no specific guidelines for the management of diabetes in acromegaly. The treatment strategy is similar to that for patients with type 2 diabetes mellitus. Metformin is the most frequently used drug as it improves insulin sensitivity and decreases the hepatic glucose output – the two most important pathophysiological factors underlying hyperglycaemia in acromegaly.9 In individuals with persistent hyperglycaemia, insulin therapy should be considered. Finally, control of growth hormone levels by surgical resection of pituitary adenoma, radiotherapy and medical therapy helps to achieve the state of euglycaemia in these individuals.10 In particular, pasireotide should be avoided in patients with acromegaly and abnormal glucose tolerance, as it has been shown to worsen the glycaemic profile due to its propensity to reduce insulin secretion and incretin hormone response.11
A literature search revealed 13 case reports of similar presentation.8,12–23 Like our case, all of them were middle-aged (20–45 years), had macroadenoma and were on insulin therapy. However, in contrast to our case, the majority of patients were male (9/13) and Caucasian (6/13); and two patients were on oral antidiabetic drugs, along with insulin. The majority of the patients in these case reports (10/13) were able to stop insulin after surgical resection of pituitary macroadenoma. Three patients did not require insulin immediately after surgery, as seen in the present case.
In conclusion, impaired glucose tolerance and diabetes mellitus are frequently associated with acromegaly. Rarely, diabetic ketoacidosis can be the presenting manifestation in these patients. Meticulous clinical examination should be performed in a patient with diabetic ketoacidosis to rule out other endocrinopathies. Control of growth hormone levels by surgical resection of somatotropinoma can help to achieve the state of euglycaemia in these individuals.