Growth hormone (GH) is approved in children to treat growth failure in several indications including GH deficiency, Turner syndrome and short children born small for gestational age,1 and is approved in the USA but not in Europe for the treatment of idiopathic short stature (ISS).2 ISS describes short children with normal GH secretion, with height >2 standard deviations (SD) below the corresponding mean height for a given age, sex, and population, in whom no identifiable disorder is present.3,4 ISS may be familial, in which the child is short compared with the reference population but remains within the range of target height based on parental height, or non-familial, in which the child is short both in comparison with the reference population and the target height based on parental height.5 GH therapy in children with ISS may be effective in reducing the deficit in height in childhood and improving adult height.3 Data on the safety of GH therapy in patients from the US with ISS suggest that the safety profile of GH is similar to that reported in other GH-treated indications and is not associated with any new safety concerns.6
The aim of this trial was to evaluate the efficacy and safety of recombinant human GH (Norditropin® NordiLet®; Novo Nordisk, Bagsværd, Denmark) therapy in paediatric patients with ISS in Korea. This prospective trial was conducted for registration purposes to seek approval of GH for the treatment of ISS in paediatric patients in Korea.
Materials and Methods
Trial design
The trial design is shown in Supplementary Figure 1. This trial was conducted in accordance with the Declaration of Helsinki7 and ICH Good Clinical Practice.8 Written informed consent was obtained from patients’ parents or legally acceptable representative before any trial-related activities. The trial was conducted at 10 sites in South Korea. For each investigational site, the adequacy of the research facility to execute the protocol requirements was confirmed by a monitor and Institutional Review Boards of each site before site initiation and any trial activities. The protocol (ClinicalTrials.gov identifier: NCT01778023; retrospectively registered 24 January 2013), the protocol amendments, the consent form, and the subject information sheet were reviewed and approved according to local regulations by the Ministry of Food and Drug Safety, and by an independent ethics committee and/or Institutional Review Board. This trial is the first of its kind for Norditropin worldwide, with the purpose of attaining a label extension for its use in the indication of ISS.
This was a 12-month, open-label, randomised, parallel-group, multicentre, interventional trial. Patients were randomised 2:1 to 12-month GH treatment period (group A) or a 6-month no treatment period followed by a 6-month GH treatment period (group B). The randomisation was performed by an external contract research organisation. The GH dosage used in this trial was 0.469 mg/kg/week, 7 days per week, administered subcutaneously in the evening (equivalent to a daily dose of 67 µg/kg/day). The GH dose selected in this trial was chosen based on the approved GH dose range for short patients born small for gestational age9 due to the similarities in growth characteristics between patients with ISS and short patients born small for gestational age.10 The sample size was determined based on a 2:1 randomisation scheme with a significance level of 5% and power of 80%, and 24 completers for group A and 12 completers for group B; on the assumption that 30% of patients would withdraw from the trial, 36 patients were randomised to group A and 18 to group B.
There were six scheduled visits to the clinic (Supplementary Figure 1). At the first visit, patients were screened for eligibility. At visit 2, occurring between 1 and 21 days after visit 1, GH treatment for group A was initiated. Patients attended three interim visits at approximately 3, 6, and 9 months (each ±7 days) after visit 2. At visit 4 (6 months after visit 2), GH treatment was initiated for group B. Visit 6, at the end of the trial, was attended approximately 12 months (±7 days) after visit 2.
Patients
To be eligible for participation in this trial, patients were required to meet all inclusion criteria and none of the exclusion criteria (Supplementary Table 1). Key inclusion criteria included pre-pubertal status (males aged ≥4 to ≤11 years and females aged ≥4 to ≤9 years): an absence of breast development in females (Tanner 1 only) and testicular volume <4 mL in males, normal thyroid function, peak GH level above 10 ng/mL following a GH stimulation test, bone age ≤12 years, and epiphyses confirmed as open in patients ≥10 years of age. For the purpose of this trial, children with height below the third percentile (based on 2007 Korean national growth charts11) and no identifiable disorder were considered as having ISS. Key exclusion criteria included a known pituitary hormone deficiency (adrenocorticotropic hormone, antidiuretic hormone, follicle-stimulating hormone, luteinising hormone, thyroid-stimulating hormone) and treatment with any GH in the 12 months before screening; and specific types of growth failure including, but not limited to, known chromosomal abnormalities associated with growth failure and altered sensitivity to GH, e.g., Turner syndrome, Noonan syndrome, Prader–Willi syndrome, chromosomal trisomies, chronic renal failure, type 1 diabetes mellitus, osteo- and chondrodystrophies, hypochondroplasia, achondroplasia, small for gestational age, chronic inflammatory states (such as inflammatory bowel disease, rheumatoid arthritis, systemic lupus, cystic fibrosis), mitochondrial myopathies, intrauterine growth retardation (defined as a birth height and weight both below the fifth percentile and not exhibiting catch-up growth by the age of 3 years), and syndromes known to be associated with growth failure.
Efficacy assessments
Height (cm) was measured in triplicate by a single observer using an identical instrument at visits 1, 2, 3, 4, 5, and 6; corresponding to screening, day 0 and months 3, 6, 9, and 12. Height data collected at visits 2 and 4 were used to calculate the primary endpoint of height velocity (Ht-V [cm/year]) after 6 months of treatment. Height data from visits 2–4 versus data from visits 4–6 were used for the secondary objective of comparing Ht-V for the first 6-month treatment period to that of the following 6-month treatment period in group A, and the exploratory objective of comparing Ht-V for the no treatment first 6-month period to that of the following 6-month treatment period in group B. In addition, height data were used to calculate the confirmatory secondary endpoint of height SD score (Ht-SDS). Ht-SDS values were calculated using the growth data from the Korea Center for Disease Control and Prevention.12 Body weight (kg) was assessed at trial entry. Parental heights were measured at visit 1 or 2. If it was not possible to obtain a parental height measurement, a reported height was substituted for a measured height. Insulin-like growth factor 1 (IGF-1) and insulin-like growth factor-binding protein 3 (IGFBP-3) were measured at visits 1, 3, 4, 5, and 6, and used to calculate the supportive secondary endpoint of change in IGF-1 and IGFBP-3 from baseline to 6 months.
Safety assessments
All treatment-emergent adverse events (AEs) reported by the patients or observed by the investigator were recorded by the investigator and evaluated further. AEs were recorded in all patients who received at least one dose of GH and were defined as any untoward event occurring in a patient independent of a causal relationship with the trial product. AEs were designated as mild (no or transient symptoms that do not interfere with daily activities), moderate (marked symptoms with moderate interference of daily activities), or severe (considerable [unacceptable] interference with daily activities). At each visit, a physical examination was performed of the head, ears, eyes, nose, throat, neck, respiratory system, cardiovascular system, gastrointestinal system (including mouth), genitourinary system, musculoskeletal system, central and peripheral nervous systems, and skin, and the findings recorded in the case report form. Abnormal findings were reported as clinically significant or not clinically significant; worsening of any clinically significant finding from the previous visit was reported as an AE. Vital signs, including diastolic and systolic blood pressure, pulse, respiratory rate, and body temperature were measured throughout the trial. At visits 1 and 3–6, clinical laboratory tests were determined for haematology, urinalysis, biochemistry, lipids, and metabolic and thyroid functions. Bone age was assessed via plain X-rays of the left hand and wrist obtained at visits 1, 4, and 6.
Statistical analysis
As defined in the trial protocol, the primary endpoint of Ht-V after 6 months of treatment was converted per 365 days for completers and last observation carried forward applied for withdrawals; the endpoint was analysed using an analysis of variance (ANOVA) model with group and sex as fixed effects, and age as a covariate. The confirmatory secondary endpoint and supportive secondary endpoints of change in IGF-1 and IGFBP-3 from baseline to 6 months were analysed using an ANOVA model with group and sex as fixed effects, and age and baseline Ht-SDS as covariates. P-values and confidence intervals (CIs) are presented together with the estimated mean effect (least square [LS] means) for each group and estimated mean group difference. The supportive secondary endpoint of Ht-V during the first 6 months and last 6 months for group A were compared using a paired t-test. An exploratory endpoint comparing Ht-V during the first and last 6 months for group B was evaluated using a paired t-test, and a 95% CI of the mean was calculated based on t-distribution.
A post hoc analysis was conducted on the primary efficacy endpoint and secondary confirmatory efficacy endpoint. This was done by repeating the analyses for all patients, but with data for patients who had height data missing at baseline replaced with their height data at screening to determine if the missing baseline height data affected the efficacy results.
Results
Patient characteristics
A total of 70 patients were screened, of whom, 16 were ineligible for inclusion as a result of failing to meet at least one of the specified inclusion criteria or for meeting at least one of the exclusion criteria (Supplementary Table 2). The remaining 54 patients were randomised 2:1 to group A (n=36) or group B (n=18). Three of the patients randomised to group B withdrew informed consent before being exposed to treatment. Baseline characteristics were similar between the two treatment groups (Table 1). At the start of the trial, the overall mean age was 6.2 years (SD 1.5), mean height was 107.1 cm (SD 8.3), and mean weight was 17.4 kg (SD 3.0).
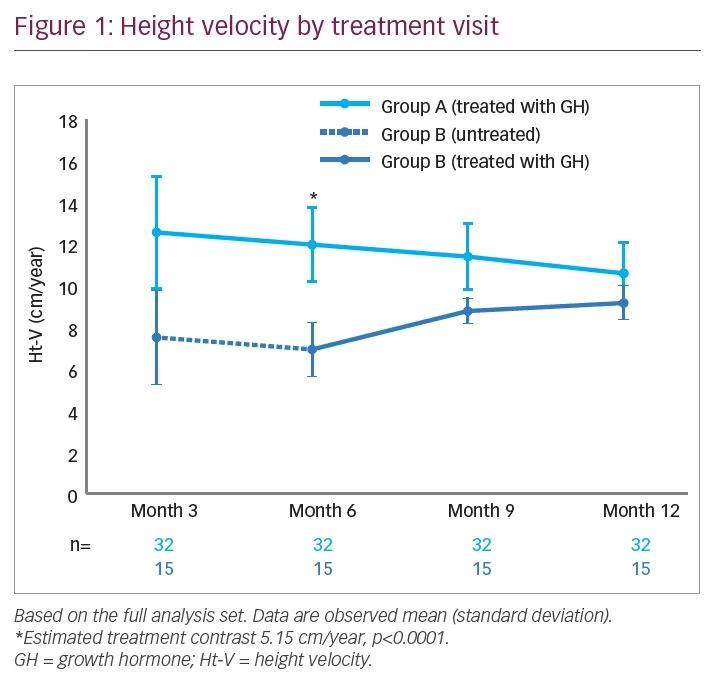
Height velocity
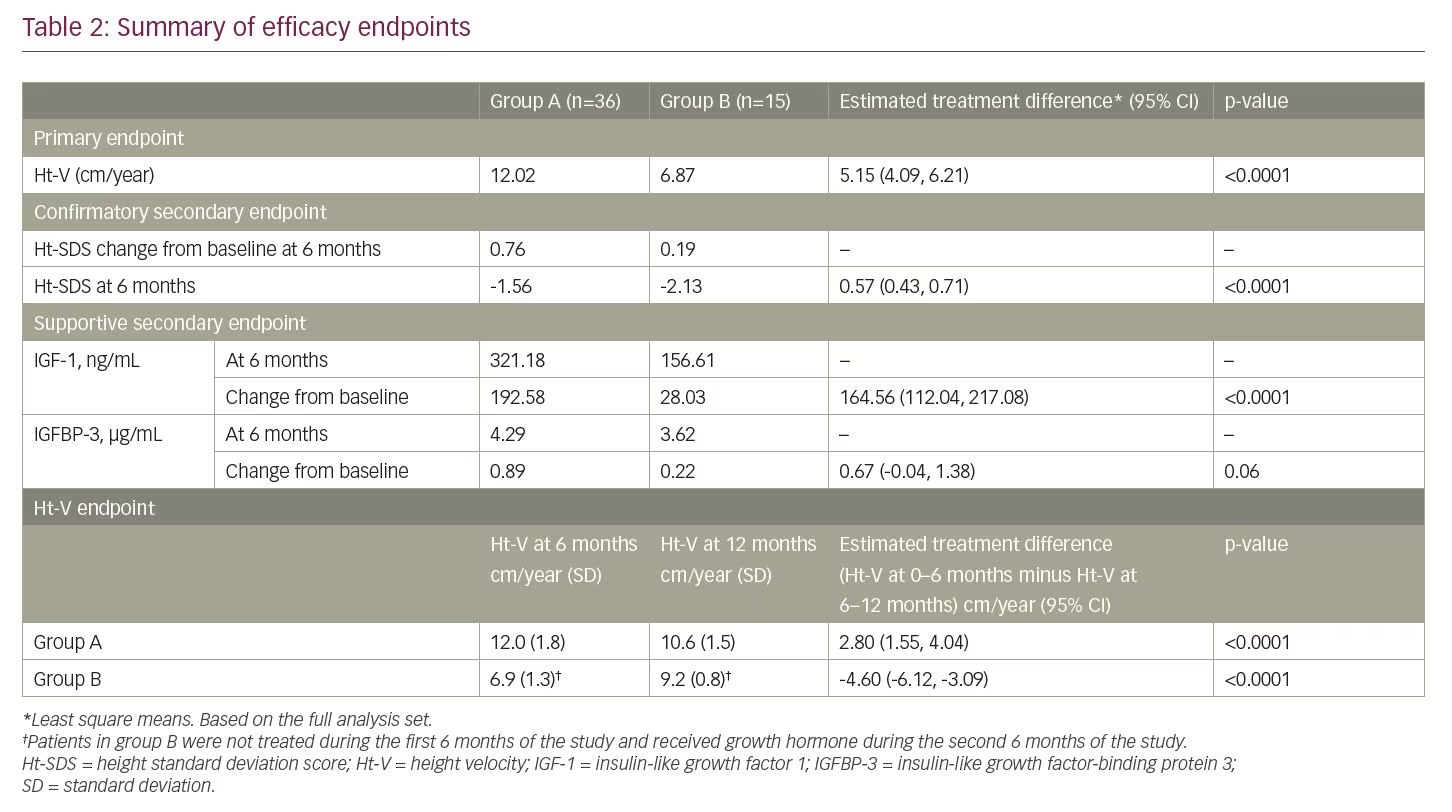
Ht-V by treatment visit is displayed in Figure 1. For the primary endpoint of Ht-V after 6 months of treatment, group A (12.02 cm/year) was higher than group B (no treatment; 6.87 cm/year), with an LS means difference of 5.15 cm/year (95% CI 4.09, 6.21; p<0.0001) (Table 2). The results of the post hoc analysis were similar to those of the primary analysis, with a mean difference between group A and group B of 5.19 cm/year (95% CI 4.09, 6.19; p<0.0001).
The mean Ht-V for group A was 12.0 cm/year (SD 1.8) and 10.6 cm/year (SD 1.5) at months 6 and 12, respectively; a comparison of Ht-V for the first and last 6 months demonstrated a statistically significant duration contrast with an LS means difference of 2.80 cm/year (95% CI 1.55, 4.04; p<0.0001). For group B, the mean Ht-V was 6.9 cm/year (SD 1.3) and 9.2 cm/year (SD 0.8) at months 6 (no treatment) and 12 (after 6 months’ GH treatment), respectively; the duration contrast between the first and last 6 months was statistically significant with an LS means difference of -4.60 cm/year (95% CI -6.12, -3.09; p<0.0001) (Table 2).
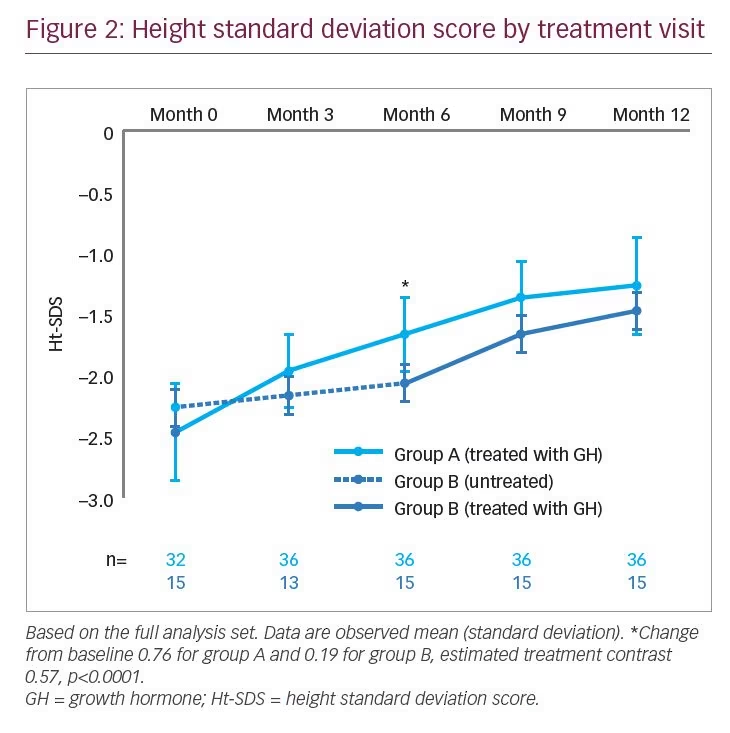
Height SDS
Ht-SDS by treatment visit is displayed in Figure 2, calculated from height data from visits 2, 4, and 6. Change in Ht-SDS (secondary endpoint) after 6 months’ GH treatment was higher for group A (0.76) than for the no-treatment group B (0.19), with an LS means difference of 0.57 (95% CI 0.43, 0.71; p<0.0001) (Table 2). A similar result was seen in the post hoc analysis, with an LS means difference of 0.57 (95% CI 0.44, 0.70; p<0.0001).
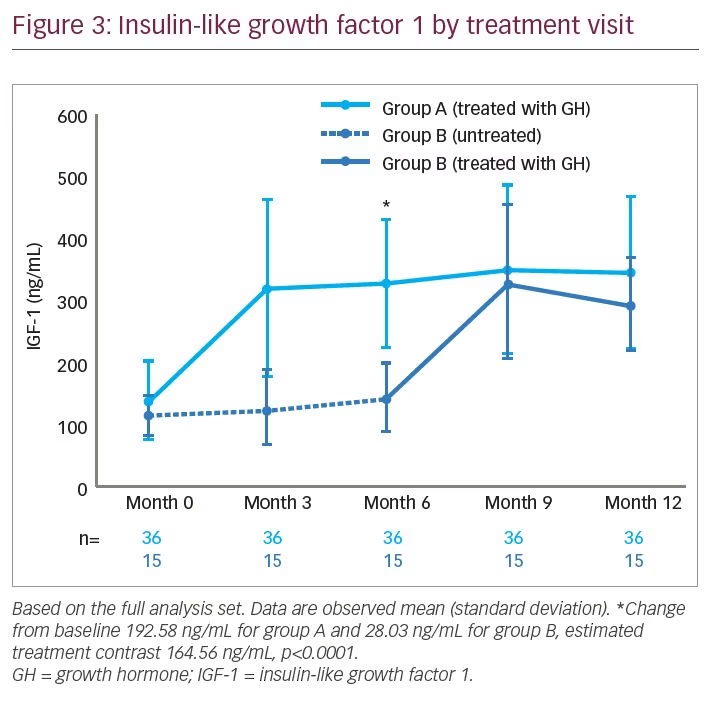
Insulin-like growth factor 1 and insulin-like growth factor-binding protein 3
IGF-1 concentration by treatment visit is shown in Figure 3. After 6 months of treatment, the change in IGF-1 concentration for group A was 192.58 ng/mL, and at the end of the 6-month no-treatment period for group B, the change in IGF-1 concentration was 28.03 ng/mL, resulting in an estimated treatment difference of 164.56 ng/mL (95% CI 112.04, 217.08; p<0.0001) (Table 2). Change in IGFBP-3 after 6 months of treatment was 0.89 µg/mL for group A and 0.22 µg/mL for group B, with an estimated treatment difference of 0.67 µg/mL (95% CI -0.04, 1.38; p=0.06) (Table 2).
Safety evaluation
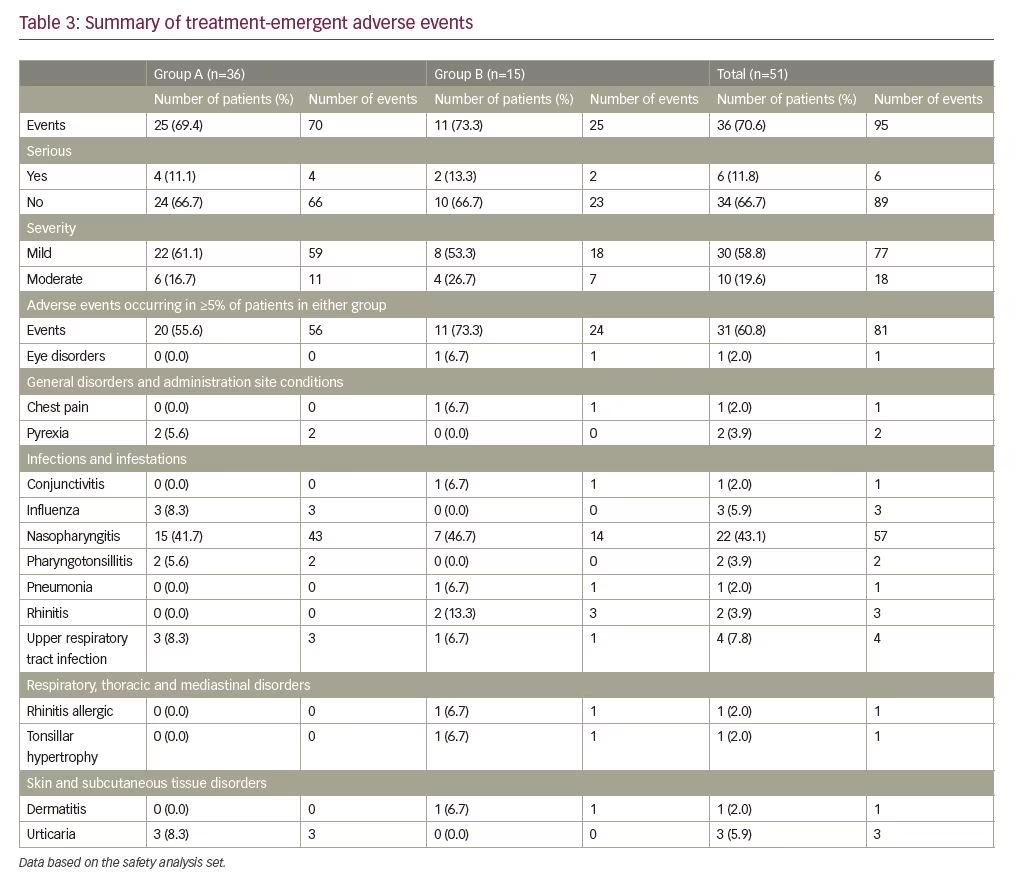
Overall, 95 AEs were reported in 36 patients during the trial (from start of GH treatment to last visit). After 12 months, a similar proportion of patients reported AEs in group A (69.4%) and group B (73.3%). Four serious AEs were reported in four patients (11.1%) in group A, and two in two patients (13.3%) in group B. In group A, there was one event each of hydrocoele, pharyngotonsillitis, tonsillar hypertrophy, and Kawasaki’s disease. In group B, there was one event of pneumonia and one event of tonsillar hypertrophy. There were no AEs leading to withdrawal in this trial. All AEs were either mild or moderate in severity (affecting 61.1% and 16.7%, respectively, of patients in group A and 53.3% and 26.7%, respectively, in group B) (Table 3). No deaths were reported. The most frequently reported AEs, occurring in at least 5% of patients in both groups, were nasopharyngitis and upper respiratory tract infections (Table 3). A total of three AEs were deemed possibly related to trial product (fatigue, contusion, and dizziness), occurring in two patients in group A.
Discussion
The results of this trial show that 6 months of GH treatment (0.469 mg/kg/week) significantly increased height in patients with ISS in Korea, as evaluated by Ht-V and Ht-SDS, compared with no treatment. In a report of 751 patients with ISS enrolled in an observational study, Ross et al.13 reported an improvement in Ht-SDS of 0.60 and 0.71 for male and female patients, respectively, following 1 year of GH therapy with an average weekly dose of 0.350 mg/kg. Patients in that study were slightly older than those in the present trial (mean age at baseline was 11.2 years [SD 3.06] versus 6.2 years [SD 1.5] in the present trial) but had a similar height deficit at treatment start (mean Ht-SDS was -2.3 [SD 0.75]).13 The lower GH dose of 0.350 mg/kg/week, in comparison to 0.469 mg/kg/week in the present trial, as well as the slightly older age at treatment start, may contribute to the difference in Ht-SDS improvements. This was reported in another study, by Albertsson-Wikland et al.,14 which showed a dose-dependent improvement in Ht-SDS, with patients receiving 0.469 mg/kg/week GH having a greater improvement in mean (SD) Ht-SDS than patients who received 0.231 mg/kg/ week (0.76 [SD 0.22] versus 0.53 [SD 0.20], respectively). In addition to GH dose, response to GH therapy has been shown to be influenced by a variety of factors, including adherence, gender, age at treatment start, and pubertal status.13,15,16
In the present trial, it was noteworthy that the gradient in Ht-SDS (Figure 2) diverged between groups for the first 6 months, but then increased in group B after month 6 when GH was initiated. From this point, the gradient for group B appeared to run approximately in parallel with that for group A. Consequently, at the end of the trial, although the Ht-SDS for group B lagged behind that of group A, demonstrating that the 6-month delay in initiating GH treatment in group B had resulted in a reduced overall growth response at 1 year, it is possible that continued treatment in group B may have allowed those patients to achieve a similar height outcome to those in group A. However, accumulated data support the potential importance of initiating treatment as early as possible to achieve optimal height outcomes, although this hypothesis requires further investigation over longer time periods. Furthermore, ISS constitutes a diverse population in which there can be varied responses to GH treatment.
Improvements in Ht-V and Ht-SDS during GH therapy were accompanied by an increase from baseline in IGF-1 and IGFBP-3, although the difference between treatment groups A and B was only significant for IGF-1. The post hoc analyses carried out on Ht-V and Ht-SDS data showed that replacement with screening data for the four patients with missing baseline height data did not affect the efficacy results.
In line with other reports of GH therapy in pre-pubertal patients with ISS,14,17 there were no unexpected AEs observed during this trial and GH treatment was generally well tolerated. Overall, the safety profiles were similar between treatment groups, with no patterns or clustering of AEs or serious AEs.
A limitation of this trial is that analyses were not performed to correct for parental height. This would account for genetic factors that may result in short stature, although such adjustments may only be appropriate in patients with parents of normal stature.18 Furthermore, the open-label design of this trial might have led to significant bias in measurement that could result in an overestimation of the effects of GH therapy on growth.
Conclusions
In conclusion, the results of this trial show that 6 months of GH treatment (0.469 mg/kg/week) significantly increased height in patients with ISS in Korea compared with 6 months of no treatment, as evaluated by Ht-V. Furthermore, GH treatment significantly increased Ht-SDS and was associated with increased IGF-1 levels compared with no treatment. No unexpected safety issues were reported and GH treatment was
well tolerated.
Supplementary Figure 1: Trial design. Graphical representation of trial design.
Supplementary Table 1: Inclusion and exclusion criteria. Documentation of full inclusion/exclusion criteria.
Supplementary Table 2: Patient disposition. Display of patient flow through the trial.