




In children, hyperthyroidism occurs less commonly compared to hypothyroidism, yet it is far more symptomatic.1 Hyperthyroidism in children is mostly due to autoimmunity, predominantly as a result of Graves’ disease.2 Non-autoimmune hyperthyroidism (NAH) is a rare cause of hyperthyroidism in children. The existence of a non-autoimmune form of goitrous hyperthyroidism was first hypothesized in 1982, affecting a family from Nancy, France.3 The affected members demonstrated symptoms of hyperthyroidism, albeit without evidence of autoimmunity. Twelve years later, the thyrotropin hormone receptor gene (TSHR) was isolated and the patients from Nancy were found to be heterozygous for V509A germline mutation. Functional study revealed V509A induced a higher basal activation of adenyl cyclase compared to the wild-type receptor, indicating activating nature of the mutation.4 Patients with activating TSHR mutation demonstrated the same classical signs and symptoms of hyperthyroidism as seen in patients with Graves’ disease. These include weight loss, hyperactivity and tachycardia. Nonetheless, subclinical hyperthyroidism among NAH cases has been described in the literature.5 To the best of our knowledge, the present report is the first to describe Malaysian cases of familial NAH (FNAH) in two different families, due to different germline TSHR-activating mutations. We noted that the same mutation exhibited variable phenotypes in both families, with regards to the age of onset and severity of hyperthyroidism.
Case reports
Family 1
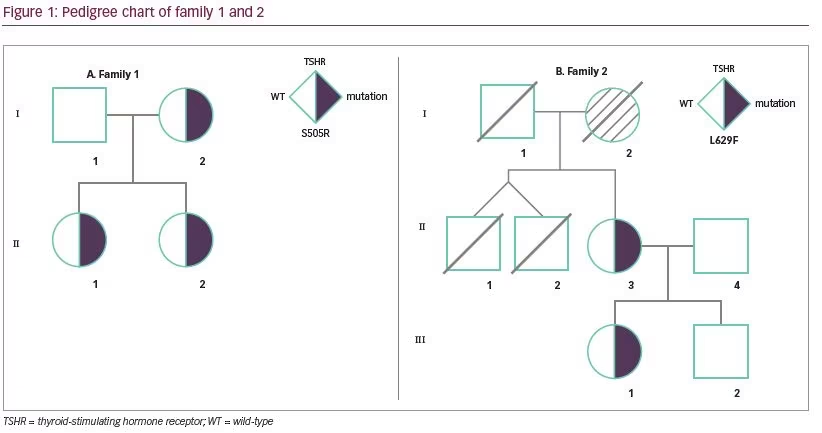
Family 1 is of Malay ethnicity with two affected female siblings (Figure 1A). In this family, possibly two generations were affected by the disorder, as the history revealed a presumptive diagnosis of Graves’ disease in their mother since teenage years.
Patient history
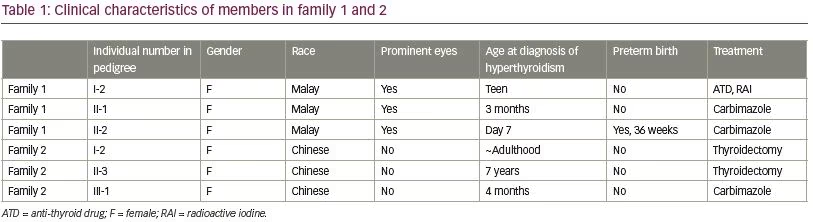
At the time of writing, the two affected girls were 7.5 and 5.0 years old (subject II-1 and II-2, respectively). History revealed that subject II-1 was under a general pediatrician follow-up since 3 months of age for persistently high free T4 (FT4) (30.0 pmol/L, normal range [NR] 10.2–27.0) and low TSH (<0.03 μIU/mL, NR 0.5–8.0), but treatment had not been started as she was asymptomatic during such time. However, she defaulted follow-up at the age of 2.0 years and only returned to care at the age of 3.5 years; at which time, which she was symptomatic for hyperthyroidism with tachycardia and hyperactivity and thus, carbimazole and propranolol were commenced.
Subject II-2 was under follow-up since day 7 of life for persistently high FT4 (50.8 pmol/L, NR 11.5–29.6) and low TSH (<0.03 μIU/mL, NR 0.5–25.0). Treatment was not started since she was asymptomatic at such time. However, at 5 months of age, her weight gain and head circumference were below the third centile, and thus, commencement of treatment was proposed. Unfortunately, the parents refused treatment at that point. They only agreed for treatment at the age of 10 months and she was started on carbimazole.
Despite treatment compliance, both siblings persistently had elevated FT4 with markedly suppressed TSH. The siblings were later referred to the pediatric endocrine clinic at the age of 5.0 years and 2.5 years, respectively. During such visitation, examination revealed both siblings had tachycardia (pulse rate 108 beats/minute and 144 beats/minute, respectively), diffused palpable goiter and mildly protruding eyes, but no true exophthalmos. Subject II-1 had FT4 of 38.9 pmol/L (NR 10.2–23.0) and TSH 0.004 μIU/mL (NR 0.5–5.0) despite being on carbimazole 5 mg twice daily (0.6 mg/kg/d). On the other hand, Subject II-2 had FT4 of 47.4 pmol/L (NR 10.2–23.0) and TSH 0.003 μIU/mL (NR 0.5–5.0) on carbimazole 5 mg once daily (0.4mg/kg/d).
Both siblings were negative for thyroid peroxidase (TPO), thyroglobulin (TG), and TSHR antibodies. Thyroid ultrasound scan showed diffuse thyroid gland enlargement in subject II-1 (right lobe 1.5 x 1.8 x 3.7 cm; left lobe 1.7 x 1.7 x 3.2 cm) and subject II-2 (1.2 x 1.4 x 3.5 cm; left lobe 1.2 x 1.4 x 3.3 cm). Ultrasound scan in both siblings did not reveal any evidence of lymphadenopathy. In view of the above findings, the siblings were then suspected to have NAH.
Family history
Further investigation on the history of the disease in the family revealed that the mother (Figure 1A; subject I-2) had a presumptive diagnosis of Graves’ disease at the age of 15 years. The mother was treated with antithyroid drugs, but it failed to control the disease symptoms satisfactorily. Thereafter, she underwent radioactive iodine ablation at the age of 24 years. She was rendered hypothyroid and was started on long term L-thyroxine replacement since then.
Mutation analysis
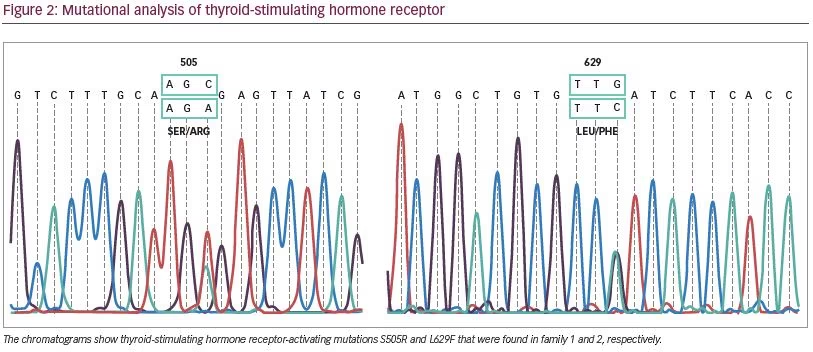
For mutational analysis, informed consent was obtained from the parents. Peripheral blood DNA was extracted from both affected siblings and the parents. Mutation analysis was carried out through direct DNA sequencing for all TSHR coding exons. This revealed a heterozygous cytosine-to-adenine transversion in exon 10 in the two siblings and the mother. This mutation leads to serine to arginine substitution, affecting TSHR codon 505 (S505R) (Figure 2). The mutation was not found in the father. Both siblings later underwent radioactive iodine ablation and are currently on L-thyroxine replacement therapy.
Family 2
The family is of Chinese ethnicity, and possibly three generations were affected by the disorder (Figure 1B). The index case (subject II-3, mother) had been treated with anti-thyroid medication since childhood for a presumptive diagnosis of Graves’ disease. The diagnosis was re-evaluated after the birth of her child (subject III-1, daughter) who had persistent hyperthyroxinemia from birth. History revealed that the mother of the index case (subject I-2, grandmother) could have had the same problem as well.
Patient history
The index case (subject II-3) presented at the age of 7 years with signs and symptoms of thyrotoxicosis. Incidentally, she had a past history of rheumatic carditis at the age of 4.5 years old resulting in chronic mitral valve regurgitation. Her cardiac function was normal and she did not require any cardiac medications. At presentation, she had a small but diffuse goiter. There were no obvious thyroid eye signs. Initial investigations showed total T4 225 nmol/L (NR 67–167), TSH 4.2 μIU/mL, while TPO and TG antibody levels were initially elevated. She was diagnosed to have autoimmune thyroiditis and was commenced on carbimazole. Follow-up serial tests showed normalization of FT4, but TSH remained persistently suppressed. During her teenage years, she had frequent relapses of thyrotoxicosis requiring increased doses of anti-thyroid medications. Her thyroid gland progressively increased in size. At the age of 20, she had an uneventful pregnancy and delivered a baby girl at term. Anti-TG and anti-TPO antibodies were re-evaluated and found to be undetectable. TSHR antibody was also undetectable. Her thyroid status was increasingly difficult to control after the delivery of her daughter, resulting in three hospital admissions for heart failure. She had total thyroidectomy at the age of 25 years followed with L-thyroxine replacement. Since then, she remained euthyroid with normal FT4 and TSH levels.
Family history
The daughter (subject III-1) of the index case was born at term via spontaneous vaginal delivery with a birth weight of 2.7 kg. She was asymptomatic and had no goiter at birth. However, she was noted to have poor weight gain, mild tachycardia of 150–160 beats/minute, persistent hyperthyroxinemia, and suppressed TSH from birth. Her serum FT4 ranged from 49 to 72 pmol/L (NR 9.14–23.81) and TSH <0.06 μIU/mL (NR 0.5–8.0). She was started on oral propranolol. However, she continued to have poor weight gain and partially controlled tachycardia until carbimazole was added at 12 months of age. Her tachycardia improved with normalized serum FT4. Her weight and height progressed satisfactorily on the tenth percentile. At age of 4 years, she developed a small, diffuse goiter. Ultrasonography showed a homogenous echogenic pattern. TSHR, TPO, and TG antibodies were undetectable. Despite normalization of FT4, the daughter’s goiter gradually increased in size with development of nodularity within the thyroid gland on serial ultrasonography. Thyroid scan showed heterogenous uptake of radio-isotope with multiple cold nodules. Total thyroidectomy was performed at the age of 12 years. A diffuse multinodular goiter weighing 60 g was removed and histopathology examination revealed macrofollicles containing colloid with no evidence of malignancy.
According to subject II-3, subject I-2 (grandmother) had a history of symptomatic hyperthyroidism associated with enlarged goiter when she was young. The grandmother underwent thyroid surgery twice—the first operation was inadequate to control her symptoms and a second operation was done to remove the thyroid gland completely. She was subsequently treated with L-thyroxine. Subject I-2 passed away at the age of 49 years. The cause of death was unknown.
Mutation analysis
For mutation analysis, informed consent was obtained and DNA from peripheral blood was isolated from subjects II-3 and III-1. All TSHR coding exons were subjected to direct DNA sequencing. This revealed heterozygous L629F (TTG>TTC) activating mutation in both of them (Figure 2).
Discussion
In the present study, we report two FNAH cases from Malaysia. The two families are unlikely to be related as they are of different ethnicities. NAH, due to activating TSHR mutations, is a rare condition. There are seven transmembrane helices (TMH) in TSHR receptor, and these helices are known to be the commonest site to harbor activating TSHR mutations. At the present time, 32 different missense changes within TMH1–7 have been reported and these resulted to gain-of-function mutation in the thyrotropin receptor.6,7 To date, 28 families with FNAH and 16 cases of sporadic NAH have been reported.8 The TSH Receptor Mutation Database (www.tsh-receptor-mutation-database.org/list.html) contains the most updated list of TSHR mutations. The database indicated a total of three familial cases have been reported from S505R5,9 and L629F10 mutations.
The daughters in both families have remained hyperthyroid since birth. The commonest cause for neonatal hyperthyroidism is secondary to maternal Graves’ disease, especially if the TSHR antibody is high during the third trimester. However, any transplacental transfer of maternal antibody should have disappeared, at the most by 4 months of postnatal age.1 Abeillon-du Payrat J et al. reported that neonatal hyperthyroidism secondary to maternal Graves’ disease generally resolves by 6 months after birth.11 However, persistence of up to 12 months has been reported in one case.12 In both families, the hyperthyroidism persisted despite antithyroid treatment. Since the cases in the two families showed undetectable thyroid autoantibodies, they were then suspected to have genetic hyperthyroidism and analyzed for TSHR mutation.
The S505R and L629F missense changes in family 1 and 2, respectively, are gain-of-function TSHR mutations. In family 1, the S505R mutation was found in three members, across two generations. The S505R mutation is located within TMH3 of TSHR protein. This mutation has been described in two previous reports.5,9 Pohlenz J et al. reported the same mutation in a 6-month-old infant who presented with weight loss and was found to have subclinical hyperthyroidism.5 Another family from Lausanne, Switzerland reported activating S505R mutation in a family in the absence of signs of autoimmunity.9
The L629F mutation was found in the second family, and the mutation is located within the TMH6 of THSR. This activating mutation has only been reported in a single FNAH case from Germany in 1997.10 The case reported L629F mutation in a 10-year-old boy with a disease onset in the neonatal period, while the mother showed childhood disease onset. This variation of disease onset is similar with both the families reported here.10
Although the same mutation was segregating among the affected members of the families, they showed varying severity of disease symptoms and presented at different stages of life (Table 1). The mothers presented at later stages of life compared to their respective children, who presented earlier in infancy. These cases illustrate the difficulty in making a diagnosis of FNAH based solely on clinical grounds especially in the second family, which was only evident after three generations. However, a similar scenario has been reported elsewhere.9
In family 2, although there was limited information regarding the grandmother’s clinical history, her history of undergoing two thyroidectomies suggested poorly controlled hyperthyroidism. The index case of family 2 (subject II-3) had clinical presentation which mimicked autoimmune hyperthyroidism, especially when the TPO and TG autoantibodies were initially elevated at the beginning. However, these two antibodies are not diagnostic for Graves’ disease and present in rather high prevalence in the general population.13,14 Besides, many of her clinical features were shared between FNAH and Graves’ disease, including the family history and prolonged usage of anti-thyroid medication to manage her disease condition. Anti-thyroid medications do not induce remission in FNAH, and lifelong treatment with these medications would be required. Therefore, a more definitive treatment is needed early on in these cases.7 On the other hand, remission rate for Graves’ disease is less than 30% after the first course of anti-thyroids for 2 years,15 and increases to only about 50% after 10 years of treatment.16
Another differentiating feature that could help detection of NAH is the lack of thyroid ophthalmopathy, which is considered to be a specific sign for Graves’ disease. However, the incidence is lesser in pediatric Graves’ disease and ranges between 25–60%.17 An absence of thyroid eye sign was initially thought to be a major marker for FNAH, only to be found out later with more reported cases, that some children did present with ocular symptoms,5 similar to our first family. In comparison, these eye signs among NAH cases typically lack the inflammatory component seen in Graves’ ophthalmopathy. The manifestation of eye signs in some families but not the others, as demonstrated in our study, illustrates the variable nature of the phenotype. We detected prominent eyes in members of the first family with S505R mutation. On the other hand, two previous reports describing cases with S505R mutation did not indicate the patients manifested any thyroid eye sign5,9 Our second family with L629F mutation did not show evidence of thyroid eye disease, unlike the family with the same mutation as reported by Fuhrer et al.10 At the present time, the pathogenesis of eye signs in NAH remains unclear. TSHR antibody, which is more specific for Graves’ disease, is probably the most helpful test to distinguish Graves’ disease from FNAH. Unfortunately, testing for TSHR antibody was not routinely done at our clinic, and was only carried out later when suspicion of NAH arose.
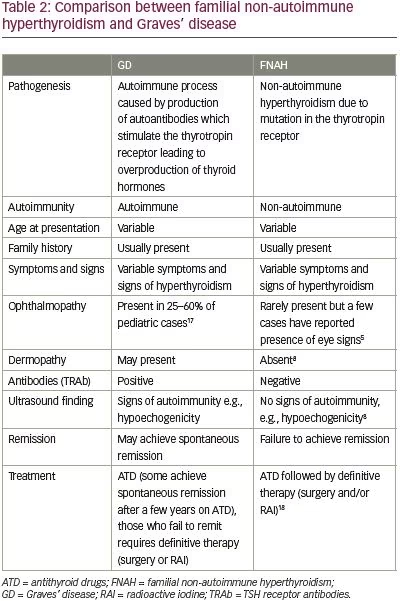
FNAH usually lacks the evidence of dermopathy as in Graves’ disease, which may aid the diagnosis.8 Furthermore, there is the absence of ultrasound signs of autoimmunity, such as hypoechogenicity and absence of lymphocytic infiltration on histology.8 Table 2 compares the diagnostic features of FNAH and Graves’ disease.
The optimal therapy for FNAH is complete ablation of thyroid tissue with total thyroidectomy followed by radioiodine administration. Anti-thyroid medication is recommended only to prepare patient for thyroid surgery, or until the patients reaches a suitable age for thyroidectomy.18 Most reported cases demonstrate the need for ablative therapy in order to better manage the disease, once the patient has become hyperthyroid.19,20 Incomplete ablation or prolonged course of anti-thyroid medications may result in frequent relapses and unwanted complications of hyperthyroidism in children, namely craniosynostosis and developmental impairment. FNAH demonstrates an autosomal dominant pattern of inheritance and thus, genetic counseling in affected families is advised. Once an index case is identified in a family, other members should be screened for the same germline mutation, including those that are asymptomatic.18
In conclusion, it is important to be aware of the presentation and natural history of genetic hyperthyroidism. The evidence of NAH in several family generations warrants THSR mutational analysis, to detect the possible genetic lesion leading to the clinical entity. Affected NAH family members having the same activating mutation may exhibit variable phenotypes with regards to the age of onset and severity of hyperthyroidism. The many similarities between the symptoms of NAH and Graves’ disease may result in a delay in reaching the correct diagnosis. Clinicians must be aware of any deviation from the most typical course of Graves’ disease, especially the lacking of autoimmune feature. An accurate diagnosis will provide patients with the most favorable outcome.


