Subclinical hypothyroidism (SH), sometimes referred to as mild hypothyroidism, is a cumbersome term used to describe the compensatory increase in thyroid-stimulating hormone (TSH) levels to preserve normal thyroid hormone levels before they fall below normal, thus averting thyroid gland failure. An unresolved issue is whether SH merits treatment when diagnosed, or whether observation is acceptable to monitor for progression to overt hypothyroidism, as there is a lack of evidence from well-designed randomised clinical trials measuring clinical outcomes.1 The objective of this review is to provide an overview of studies that have investigated the association of SH with an elevated cardiovascular disease (CVD) risk.
Is there a link between subclinical hypothyroidism and
cardiovascular disease risk?
A series of longitudinal population studies have found that SH is associated with a higher risk of CVD, and this was confirmed in a large meta-analysis published in 2010.2 The extent of the CVD risk was proportional to the degree of the TSH elevation. The effect was not related to traditional risk factors such as obesity, hypertension or dyslipidemia, and it was not related to the underlying autoimmune dysfunction that underlies the most common cause of SH, Hashimoto’s thyroiditis.3 No effect of age was noted in this meta-analysis,2 although an earlier population study suggested that the elderly may have reduced CVD risk with SH.4 A higher risk of stroke, another major vascular disease, with SH was observed in subjects younger than 65 and those with higher TSH concentrations.5
What mediates the higher cardiovascular disease risk of
subclinical hypothyroidism?
SH is a pro-inflammatory and pro-coagulant state. Elevations in interleukin (IL)-6 and C-reactive protein (CRP) have been detected in SH patients.6,7 Metabolic effects include higher levels of free fatty acids (FFA), which can also lead to inflammatory cellular actions.8 SH patients also display platelet hyper-reactivity.9 Endothelium-dependent relaxation is also impaired in these patients,6,7 and an altered pattern of endothelial-derived microparticles is associated with SH in patients with heart failure.10 A recent meta-analysis addressed the controversy about whether lipid profiles are altered in SH, and found evidence of higher serum total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), and triglycerides.11 In a small intervention study, thyroxine treatment of subjects with SH reduced the elevated levels of serum total cholesterol and LDL-C.12
Are these alterations mediated by extra-thyroidal
thyroid-stimulating hormone action?
To explore whether TSH is capable of inducing such responses, investigators have studied thyroid cancer survivors who have been treated with thyroidectomy and radioiodine ablation of any remnant tissue, and who are on treatment with levothyroxine. Some of these patients undergo exogenous TSH administration to screen for early evidence of thyroid cancer recurrence by determining if stimulated thyroglobulin blood levels are high or not. For these few days, TSH levels rise acutely and are quite elevated, whereas thyroid hormone levels remain unchanged. This setting provides researchers with an interesting model to study the extra-thyroidal effects of acute TSH elevations in vivo (i.e., no thyroid gland). It turns out that acute TSH stimulation leads to many of the same responses seen in SH. These include impaired endotheliumdependent vasodilation, higher platelet reactivity, elevations in FFA, CRP, and IL-6.13–16 Other acute responses include rises in tumour necrosis factor (TNF) a, leptin, lipoperoxide and microparticles.13,17,18
Which extra-thyroidal sites are targeted by thyroid-stimulating hormone to cause the subclinical hyperthyroidism-associated alterations?
The compensatory elevated level of TSH is beneficial in overcoming partial thyroid failure to preserve thyroid function in SH. However, the same TSH elevation may disrupt the functions of TSH receptor (TSHR)- expressing non-thyrocyte cells. We have an incomplete understanding of this evolving research area. It is increasingly recognised that TSHR expression is not restricted to thyrocytes.19–22 This might explain the abnormalities seen in SH.
The most studied cell type in this regard is the adipocyte, with studies going back several decades showing that TSH was one of several ligands capable of stimulating G proteins in rodent adipocytes.23 An adiposeselective conditional deletion of the TSHR in mice caused adipocyte hypertrophy, higher basal lipolysis and a reduced lipolytic response to exogenous TSH.24 TSHR expression has been documented in human adipocytes,25,26 with higher expression in the neonatal/paediatric age range.27 TSH-stimulated human adipocytes undergo lipolysis, increase production and release of IL-6 and MCP-1 in vitro, and are more resistant to insulin.15,16,28–31 These responses are consistent with the pattern seen in SH and in the acute TSH stimulation setting of thyroid cancer patients. The factors they release could also indirectly contribute to the abnormalities in endothelial and platelet function in these conditions.
Recently, it was suggested TSH may have an anti-lipolytic/pro-lipogenic effect. TSH decreased adipose triglyceride lipase (ATGL) expression,32 and increased TG accumulation due to induction of glycerol 3-phosphate acyltransferase (GPAT) 3, the rate-limiting step for TG synthesis.33 These results are at odds with findings of several other groups, including lipolytic effects of TSH on human adipocytes in culture,15,28 elevated FFA levels that occur in patients with subclinical hypothyroidism8 and following rhTSH administration in vivo,15 that expression of TSHR in adipocytes in mice induces lipolysis in vivo,34 and TSH inhibiting expression of the lipogenic enzyme fatty acid synthase (FAS).35
TSHR expression has also been observed in human aortic endothelial cells, which increased NO production is response to TSH.36 Using human microvascular endothelial cells, TSH was shown to stimulate angiogenesis and proliferation.37 Upregulation in intercellular adhesion molecules, and down-regulation of prostacyclin and eNOS was induced by TSH in human umbilical vein endothelial cells.38 Therefore, it is possible that direct action of high TSH levels on the endothelium could be responsible for the endothelial dysfunction in SH.
Another vascular target could be vascular smooth muscle cells. They have been reported to express TSHR at the mRNA level,39 but others
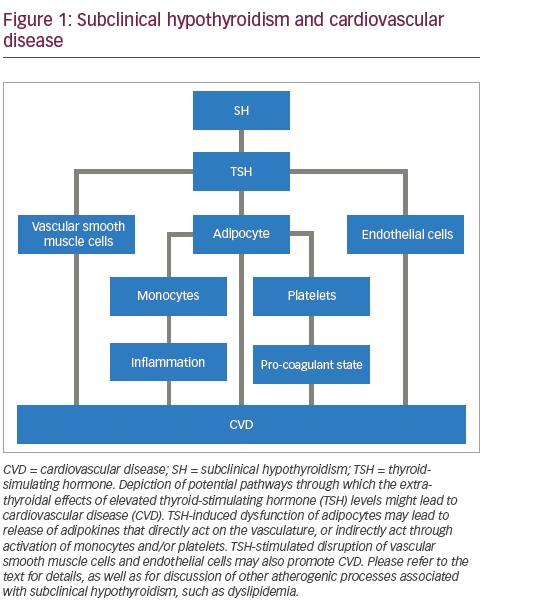
failed to confirm this expression, perhaps due to low abundance of the receptor.40 TSH stimulation of human coronary artery smooth muscle cells does lead to cAMP elevations, and human umbilical artery smooth muscle cells are induced to proliferate upon TSH treatment in vitro.41,42
Hepatocytes have been identified as expressing TSHR.43 TSH increases 3-hydroxy-3-methylglutaryl-coenzyme A (HMG CoA) reductase expression and activity, suggesting a link to cholesterol hepatic synthesis.44 TSH also decreases an 5’ AMP-activated protein kinasemediated inhibitory phosphorylation of HMG CoA reductase, further promoting higher HMG CoA reductase activity.45 In addition, TSH increases sterol regulatory element-binding protein 1 (SREBP-1) action, promoting hepatic lipogenesis,46 and inhibits bile acid formation.47
Other extra-thyroidal sites of TSHR expression include the retro-orbital tissue and bone. However, it is not likely that TSH action at those sites would induce responses that would predispose to cardiovascular risk.
Concluding statement
Cardiovascular disease is a major cause of morbidity and mortality. Many modifiable risk factors have been identified for CVD, and targeting these risk factors can prevent CVD.48 SH, a lesser-known predisposing condition, deserves more attention in the strategy to reduce CVD.49 It is heartening to know that a randomised controlled trial called the Thyroid hormone Replacement for Untreated older adults with Subclinical hypothyroidism a randomised placebo-controlled Trial among older adults (NCT01660126) is finally underway. It is hoped that within the next few years we will know much more regarding the best approach to SH and the prevention of CVD.